Get your patient on Blujepa - Gepotidacin tablet, Film Coated (Gepotidacin)
Blujepa - Gepotidacin tablet, Film Coated prescribing information
INDICATIONS AND USAGE
BLUJEPA is a triazaacenaphthylene bacterial type II topoisomerase inhibitor indicated for the treatment of the following infections caused by susceptible microorganisms:
- Uncomplicated urinary tract infections (uUTI) in female adult and pediatric patients 12 years of age and older weighing at least 40 kilograms (kg). (1.1 )
- Uncomplicated urogenital gonorrhea in adult and pediatric patients 12 years of age and older weighing at least 45 kilograms who have limited or no alternative treatment options. Approval of this indication is based on limited clinical safety data for this indication. (1.2 , 6.1 )
Usage to Reduce Development of Drug-Resistant Bacteria
To reduce the development of drug-resistant bacteria and maintain the effectiveness of BLUJEPA and other antibacterial drugs, BLUJEPA should be used only to treat infections that are proven or strongly suspected to be caused by bacteria. (1.3 )
Treatment of Uncomplicated Urinary Tract Infections
BLUJEPA is indicated in female adult and pediatric patients 12 years of age and older weighing at least 40 kilograms (kg) for the treatment of uncomplicated urinary tract infections (uUTI) caused by the following susceptible microorganisms: Escherichia coli , Klebsiella pneumoniae , Citrobacter freundii complex, Staphylococcus saprophyticus , and Enterococcus faecalis .
1.2 Treatment of Uncomplicated Urogenital Gonorrhea
BLUJEPA is indicated in adult and pediatric patients 12 years of age and older weighing at least 45 kilograms (kg) who have limited or no alternative options for the treatment of uncomplicated urogenital gonorrhea caused by susceptible strains of Neisseria gonorrhoeae. Approval of this indication is based on limited clinical safety data for BLUJEPA [see Adverse Reactions (6.1 )].
Usage to Reduce Development of Drug-Resistant Bacteria
To reduce the development of drug-resistant bacteria and maintain the effectiveness of BLUJEPA and other antibacterial drugs, BLUJEPA should be used only to treat infections that are proven or strongly suspected to be caused by susceptible bacteria. When culture and susceptibility information are available, they should be considered in selecting or modifying antibacterial therapy. In the absence of such data, local epidemiology and susceptibility patterns may contribute to the empiric selection of therapy.
DOSAGE AND ADMINISTRATION
- uUTI: The recommended dosage of BLUJEPA is 1,500 mg (two 750 mg tablets) taken orally, twice daily (approximately 12 hours apart), for 5 days. (2.1 )
- Uncomplicated Urogenital Gonorrhea: The recommended dosage of BLUJEPA is 3,000 mg (four 750 mg tablets) taken orally, followed by a second dose of 3,000 mg (four 750 mg tablets) approximately 12 hours later. (2.2 )
- Administer BLUJEPA tablets after a meal to reduce the possibility of gastrointestinal intolerance. (2.3 )
Recommended Dosage for Female Adult and Pediatric Patients 12 Years of Age and Older Weighing at Least 40 kg for Uncomplicated UTI
The recommended dosage of BLUJEPA is 1,500 mg (two 750 mg tablets) taken orally, twice daily (approximately 12 hours apart) for 5 days in female adult and pediatric patients 12 years of age and older with uncomplicated uUTI [see Dosage and Administration (2.3 )] .
2.2 Recommended Dosage for Adult and Pediatric Patients 12 Years of Age and Older Weighing at Least 45 kg for Uncomplicated Urogenital Gonorrhea
The recommended dose of BLUJEPA is 3,000 mg (four 750 mg tablets) taken orally, followed by a second dose of 3,000 mg (four 750 mg tablets) approximately 12 hours later in adult and pediatric patients 12 years of age and older weighing at least 45 kg with uncomplicated urogenital gonorrhea [see Dosage and Administration (2.3 )].
Do not increase the dose, extend the duration of treatment, or reduce the interval between doses due to the risk of QTc interval prolongation [see Warnings and Precautions (5.1 )].
Important Administration Instructions
Recommendations Regarding Missed Dose(s)
If a dose is missed, instruct patients to take the missed dose as soon as possible. For uUTI, do not double the dose to make up for a missed dose.
DOSAGE FORMS AND STRENGTHS
Each film coated tablet of BLUJEPA contains 750 mg of gepotidacin. BLUJEPA tablets are yellow, capsule shaped, and debossed with “GS GU3” on one side and plain on the other side.
USE IN SPECIFIC POPULATIONS
Pregnancy
Pregnancy Exposure Registry
A pregnancy exposure registry will be established to monitor pregnancy outcomes in women exposed to BLUJEPA during pregnancy. Pregnant women exposed to BLUJEPA, and healthcare providers are encouraged to contact GlaxoSmithKline at 1‑888‑825‑5249.
Risk Summary
There are no available data on the use of BLUJEPA in pregnant women to evaluate for a drug‑associated risk for major birth defects, miscarriage, or other adverse maternal or fetal outcomes.
In embryo‑fetal development studies in mice and rats, decreased fetal weights and increased fetal mortality (late resorptions) were observed at exposures less than the maximum recommended human dose (MRHD). In a mouse pre- and postnatal development study, there were no adverse developmental effects at exposures approximately equal to the MRHD (see Data ) .
The background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage, in clinically recognized pregnancies, is 2% to 4% and 15% to 20%, respectively.
Data
Animal Data: Gepotidacin did not cause malformations when orally administered in embryo‑fetal development studies during organogenesis. Decreased fetal weights were seen in rats dosed orally with 450 mg/kg/day or greater (less than the MRHD based on AUC extrapolated from nonpregnant rats). Decreased fetal weights and increased late fetal resorptions were seen in mice at oral doses 500 mg/kg/day or greater (less than the MRHD based on AUC extrapolated from nonpregnant mice).
In a pre- and post‑natal development study in mice given oral doses of up to 1,000 mg/kg/day (approximately equal to the MRHD), there were no gepotidacin effects on parturition, or post‑natal growth and development of the offspring.
Lactation
Risk Summary
There are no data on the presence of gepotidacin in human milk, its effects on the breastfed child, or on milk production. Based on a study in lactating mice, gepotidacin is likely transferred into milk (see Data ). When a drug is present in animal milk, it is likely that the drug will be present in human milk.
The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for BLUJEPA and any potential adverse effects on the breastfed child from BLUJEPA or from the underlying maternal condition.
Data
Gepotidacin concentrations in milk have not been measured directly in animals. However, in a pre- and post‑natal development study when gepotidacin was orally administered to maternal mice during pregnancy and lactation, at all tested doses, gepotidacin was detected in the plasma of their nursing offspring (with dosing 200 mg/kg/day or greater, which was expected to result in maternal exposures as low as 0.2-times the MHRD as extrapolated from exposures in nonpregnant mice).
Pediatric Use
Uncomplicated UTI
The safety and effectiveness of BLUJEPA for the treatment of uUTI have been established for the treatment of uncomplicated UTI in female pediatric patients 12 years of age and older, weighing at least 40 kg. Use of BLUJEPA in these patients is supported by evidence from adequate and well‑controlled studies of BLUJEPA in female adult and pediatric patients 12 years of age and older with uUTI and additional pharmacokinetic data in pediatric patients (12 to <18 years of age) [see Clinical Pharmacology (12.3 ), Clinical Studies (14.1 )] . The safety profile of BLUJEPA in female pediatric patients 12 years of age and older was similar to female adults with uUTI treated with BLUJEPA [see Adverse Reactions (6.1 ), Clinical Studies (14.1 )] .
The safety and effectiveness of BLUJEPA have not been established in pediatric patients less than 12 years of age or weighing less than 40 kg.
Uncomplicated Urogenital Gonorrhea
The safety and effectiveness of BLUJEPA for the treatment of uncomplicated urogenital gonorrhea have been established in pediatric patients 12 years of age and older weighing at least 45 kg who have limited or no alternative treatment options [see Indications and Usage (1.1 )] . Use of BLUJEPA in these patients is supported by evidence from an adequate and well‑controlled study (Trial 3) in adult and pediatric patients with uncomplicated urogenital gonorrhea weighing at least 45 kg, and additional pharmacokinetic analyses showing similar drug exposure levels for pediatric patients (12 to <18 years of age) compared with adults [see Clinical Pharmacology (12.3 ), Clinical Studies (14.2 )] . The safety profile of BLUJEPA was similar between pediatric healthy volunteers 12 to <18 years of age (n = 12) who received two 3,000 mg doses compared to adults [see Adverse Reactions (6.1 )] .
The safety and effectiveness of BLUJEPA have not been established for the treatment of uncomplicated urogenital gonorrhea in pediatric patients less than 12 years of age or weighing less than 45 kg.
Geriatric Use
Uncomplicated UTI
Of the total number of patients who received treatment with BLUJEPA in the uUTI studies (Trials 1 and 2), 226 (14%) were 65 to less than 75 years of age and 127 (8%) were 75 years of age and older [see Clinical Studies (14.1 )] . No overall differences in safety or effectiveness of BLUJEPA were observed between patients 65 years of age and older and younger adult patients, but greater sensitivity of some older individuals cannot be ruled out.
BLUJEPA is known to be substantially excreted by the kidney, and the risk of adverse reactions to this drug may be greater in patients with impaired renal function [see Warnings and Precautions (5.1 ), Use in Specific Populations (8.6 ), Clinical Pharmacology (12.3 )] .
Uncomplicated Urogenital Gonorrhea
Trial 3 did not include sufficient numbers of subjects aged 65 years and older to determine whether they respond differently from younger subjects.
Renal Impairment
For Patients with uUTI or Uncomplicated Urogenital Gonorrhea
No dosage adjustment is required in patients with mild renal impairment (eGFR 60 to 89 mL/min) or moderate renal impairment (eGFR 30 to 59 mL/min).
Avoid use of BLUJEPA in patients with severe renal impairment or kidney failure (eGFR <30 mL/min), including those receiving dialysis, due to increased exposure to gepotidacin and the risk of QTc prolongation [see Warnings and Precautions (5.1 ), Clinical Pharmacology (12.3 )] .
Additional Recommendations for Patients with Uncomplicated Urogenital Gonorrhea
For patients with uncomplicated urogenital gonorrhea and moderate renal impairment (eGFR 30 to 59 mL/min), avoid use of BLUJEPA when additional risk factors for increased gepotidacin exposure are present [see Warnings and Precautions (5.1 )] .
Hepatic Impairment
For Patients with uUTI or Uncomplicated Urogenital Gonorrhea
No dosage adjustment is required in patients with mild or moderate hepatic impairment (Child‑Pugh Class A/B).
Avoid use of BLUJEPA in patients with severe hepatic impairment (Child‑Pugh Class C) due to increased exposure to gepotidacin and the risk of QTc prolongation [see Warnings and Precautions (5.1 ), Clinical Pharmacology (12.3 )] .
Additional Recommendations for Patients with Uncomplicated Urogenital Gonorrhea
For patients with uncomplicated urogenital gonorrhea and moderate hepatic impairment (Child-Pugh Class B), avoid use of BLUJEPA when additional risk factors for increased gepotidacin exposure are present [see Warnings and Precautions (5.1 ) and Clinical Pharmacology (12.3 )] .
WARNINGS AND PRECAUTIONS
- QTc Prolongation:
- Avoid use of BLUJEPA in patients with a history of QTc prolongation, or with relevant pre‑existing cardiac disease, or in patients receiving drugs that prolong the QTc interval. (5.1 )
- Due to an increase in gepotidacin exposure and the risk of QTc interval prolongation, avoid use of BLUJEPA in patients who have any of the following risk factors: (5.1 , 7.1 , 8.6 , 8.7 )
- Concomitant use of strong CYP3A4 inhibitors
- Severe renal impairment (estimated glomerular filtration rate [eGFR] <30 mL/min)
- Severe hepatic impairment (Child-Pugh Class C)
- Additionally, avoid BLUJEPA in uncomplicated urogenital gonorrhea patients, who have any of the following risk factors for increased gepotidacin exposure: (5.1 , 7.1 , 8.6 , 8.7 )
- Concomitant use of moderate CYP3A4 inhibitors
- Two or more of the following risk factors: Body weight between 45 kilograms and 60 kilograms, Moderate renal impairment (eGFR 30 to 59 mL/min), Moderate hepatic impairment (Child-Pugh Class B)
- Acetylcholinesterase Inhibition: Dysarthria and other adverse reactions have been reported in patients receiving BLUJEPA. Monitor patients with underlying medical conditions that may be exacerbated by acetylcholinesterase inhibition and patients receiving succinylcholine‑type neuromuscular blocking agents, systemic anticholinergic medications, or non‑depolarizing neuromuscular blocking agents. (5.2 )
- Hypersensitivity Reactions: Hypersensitivity reactions, including anaphylaxis, have been reported in patients receiving BLUJEPA. If an allergic reaction to BLUJEPA occurs, discontinue the drug and institute appropriate supportive measures. (5.3 )
- Clostridioides difficile Infection (CDI): CDI has been reported with nearly all systemic antibacterial agents, including BLUJEPA. Evaluate patients who develop diarrhea. (5.4 )
5.1 QTc Prolongation
A dose and concentration-dependent prolongation of the QTc interval has been observed with BLUJEPA [see Clinical Pharmacology (12.2 )].
Avoid BLUJEPA in patients with a history of QTc interval prolongation or those with relevant pre ‑ existing cardiac disease, patients taking antiarrhythmic agents, or other medications that may potentially prolong the QTc interval [ see Drug Interactions (7.4 )].
Due to an increase in gepotidacin exposure (C max ), and the risk of QTc interval prolongation, avoid BLUJEPA in patients who have any of the following risk factors [see Drug Interactions (7.1 ), Use in Specific Populations (8.6 , 8.7 )]:
- Concomitant use of strong CYP3A4 inhibitors
- Severe renal impairment (estimated glomerular filtration rate [eGFR] <30 mL/min)
- Severe hepatic impairment (Child-Pugh Class C)
Additionally, avoid BLUJEPA in uncomplicated urogenital gonorrhea patients who also have any of the following risk factors for increased gepotidacin exposure [see Drug Interactions (7.1 ), Use in Specific Populations (8.6 , 8.7 )]:
- Concomitant use of moderate CYP3A4 inhibitors
- Two or more of the following risk factors:
- Body weight between 45 kg and 60 kg
- Moderate renal impairment (eGFR 30 to 59 mL/min)
- Moderate hepatic impairment (Child ‑ Pugh Class B)
If administration of BLUJEPA cannot be avoided in these patients, monitor and correct serum electrolyte abnormalities and collect an ECG prior to administration and during treatment, as clinically indicated.
Acetylcholinesterase Inhibition
BLUJEPA is a reversible acetylcholinesterase inhibitor in in vitro laboratory studies. Adverse reactions including dysarthria, syncope, presyncope, muscle spasms, diarrhea, nausea, vomiting, abdominal pain, hypersalivation, and hyperhidrosis which are potentially attributed to acetylcholinesterase inhibition, have been observed in clinical trials [see Adverse Reactions (6.1 )] . Increased cholinergic effects can be associated with severe adverse reactions including atrioventricular block, bradycardia, bronchospasm, and seizures/convulsions. Monitor patients with medical conditions that may be exacerbated by acetylcholinesterase inhibition.
BLUJEPA, as an acetylcholinesterase inhibitor, may exaggerate the neuromuscular effects of succinylcholine‑type muscle relaxation during anesthesia. BLUJEPA may exaggerate the effects of other acetylcholinesterase inhibitors. Monitor patients for exaggerated neuromuscular blockade or excessive cholinergic effects.
Because BLUJEPA may antagonize the effects of systemic anticholinergic medications or non‑depolarizing neuromuscular blocking agents, monitor patients if BLUJEPA is concomitantly administered with these medications [see Drug Interactions (7.3 )] .
Hypersensitivity Reactions
Hypersensitivity reactions, including anaphylaxis, have been reported in patients receiving BLUJEPA [see Adverse Reactions (6.1 )] . BLUJEPA is contraindicated in patients with a history of severe hypersensitivity to BLUJEPA [see Contraindications (4 )]. Before therapy with BLUJEPA is instituted, carefully inquire about previous hypersensitivity reactions to BLUJEPA. If an allergic reaction to BLUJEPA occurs, discontinue the drug and institute appropriate supportive measures.
Clostridioides difficile Infection
Clostridioides difficile (C. difficile) infection (CDI) has been reported for nearly all systemic antibacterial agents, including BLUJEPA, and may range in severity from mild diarrhea to fatal colitis [see Adverse Reactions (6 )] . Treatment with antibacterial agents alters the normal flora of the colon leading to overgrowth of C. difficile .
C. difficile produces toxins A and B which contribute to the development of CDI. Hypertoxin producing isolates of C. difficile cause increased morbidity and mortality, as these infections can be refractory to antimicrobial therapy and may require colectomy. CDI must be considered in all patients who present with diarrhea following antibacterial drug use. Careful medical history is necessary since CDI has been reported to occur over 2 months after the administration of antibacterial agents.
If CDI is suspected or confirmed, ongoing antibacterial drug use not directed against C. difficile may need to be discontinued. Appropriate fluid and electrolyte management, protein supplementation, antibacterial drug treatment of C. difficile , and surgical evaluation should be instituted as clinically indicated.
Development of Drug-Resistant Bacteria
Prescribing BLUJEPA in the absence of a proven or strongly suspected bacterial infection is unlikely to provide benefit to the patient and increases the risk of the development of drug‑resistant bacteria [see Indications and Usage (1.3 )] .
ADVERSE REACTIONS
The following adverse reactions are discussed in greater detail in other sections of the labeling:
Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared with rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Clinical Trial Experience in Patients with Uncomplicated UTI
The safety of BLUJEPA was evaluated in 2 double‑blind, active‑controlled, randomized trials in female adult and pediatric patients 12 years of age and older with uUTI (Trial 1 and Trial 2). A total of 1,570 patients were treated with BLUJEPA and 1,558 patients were treated with nitrofurantoin (pooled safety populations for BLUJEPA and nitrofurantoin, respectively). Patients received treatment for a median duration of 5 days.
In Trials 1 and 2 (pooled, intent-to-treat [ITT] population), the median age of patients treated with BLUJEPA was 49 (range 13 to 89) years; <1% were <18 years, 77% of patients were 18 to 64 years, 14% were 65 to 74 years, and 8% were ≥75 years. Patients were female (100%) and White (83%), Black or African American (7%), Asian (5%), or American Indian or Alaskan Native (4%); for ethnicity, 33% identified as Hispanic/Latino and 67% as non-Hispanic/Latino. The majority of patients were enrolled from the U.S. (55%).
Serious Adverse Reactions and Adverse Reactions Leading to Discontinuation: In the pooled trials (Trials 1 and 2), serious adverse reactions occurred in 1/1,570 (<1%) uUTI patients treated with BLUJEPA and 1/1,558 (<1%) uUTI patients treated with nitrofurantoin. The serious adverse reaction reported with BLUJEPA was dysarthria. No adverse reaction led to death in either treatment group.
In the pooled trials, adverse reactions leading to discontinuation of treatment occurred in 79/1,570 (5%) uUTI patients treated with BLUJEPA and 30/1,558 (2%) uUTI patients treated with nitrofurantoin. Adverse reactions occurring in >1% of patients leading to treatment discontinuation in patients treated with BLUJEPA included diarrhea (3%) and nausea (1%).
Common Adverse Reactions: Table 1 lists the adverse reactions occurring in ≥1% of uUTI patients receiving BLUJEPA in the pooled trials (Trials 1 and 2).
| a Abdominal pain includes abdominal pain, abdominal pain upper, abdominal pain lower, abdominal tenderness, abdominal discomfort, and gastrointestinal pain. | ||
Adverse Reaction | BLUJEPA N = 1,570 n (%) | Nitrofurantoin N = 1,558 n (%) |
Diarrhea | 258 (16) | 51 (3) |
Nausea | 146 (9) | 64 (4) |
Abdominal pain a | 60 (4) | 34 (2) |
Flatulence | 43 (3) | 8 (<1) |
Headache | 38 (2) | 40 (3) |
Soft feces | 37 (2) | 8 (<1) |
Dizziness | 29 (2) | 19 (1) |
Vomiting | 28 (2) | 10 (<1) |
Vulvovaginal candidiasis | 20 (1) | 18 (1) |
Diarrhea: In Trials 1 and 2, diarrhea was reported in 258/1,570 (16%) uUTI patients receiving BLUJEPA; 11% mild, 5% moderate, and <1% severe. The diarrhea started within the first 2 days of treatment for the majority of patients and the median duration of diarrhea was 4 days.
Adverse Reactions Occurring in Less than 1% of uUTI Patients Receiving BLUJEPA in Trials 1 and 2 (Pooled):
Gastrointestinal Disorders: Abdominal distension, dyspepsia (includes epigastric discomfort, eructation)
Nervous System Disorders: Presyncope, dysarthria
Infections and Infestations: Clostridioides difficile infection
Musculoskeletal and Connective Tissue Disorders: Muscle spasms
Vascular Disorders: Hot flush
Cardiac Disorders: Tachycardia
Eye Disorders: Blurred vision
Ear and Labyrinth Disorders: Vertigo
General Disorders and Administration Site Disorders: Fatigue
Investigations: Alanine aminotransferase/aspartate aminotransferase increased
Skin and Subcutaneous Tissue: Rash, hyperhidrosis
Immune System Disorders: Hypersensitivity reactions
Select Adverse Reactions Occurring in uUTI Patients Receiving BLUJEPA in Phase 1 and 2 Clinical Studies: Gastrointestinal Disorders: Hypersalivation (with oral daily doses ranging from 100 mg to 6,000 mg, which includes not approved doses)
Clinical Trial Experience in Patients with Uncomplicated Urogenital Gonorrhea
The safety of BLUJEPA was evaluated in a randomized, active‑controlled trial (Trial 3) comparing BLUJEPA to ceftriaxone and azithromycin in adult and pediatric patients 12 years of age and older with uncomplicated urogenital gonorrhea. A total of 309 patients received at least one dose of BLUJEPA (safety population).
In Trial 3 (ITT population), the median age of patients randomized to receive BLUJEPA was 33 (range 16 to 64) years; >99% of patients were 18 to 65 years (no patients were >65 years). Overall, patients randomized to receive BLUJEPA treatment were predominately male (89%) and White (74%).
Serious Adverse Reactions and Adverse Reactions Leading to Discontinuation: No serious adverse reactions occurred in uncomplicated urogenital gonorrhea patients treated with BLUJEPA in Trial 3. Adverse reactions leading to discontinuation of treatment occurred in 3/309 (<1%) of uncomplicated urogenital gonorrhea patients treated with BLUJEPA.
Common Adverse Reactions: Table 2 lists the adverse reactions occurring in ≥2% of uncomplicated urogenital gonorrhea patients receiving BLUJEPA in Trial 3.
| a Abdominal pain includes abdominal pain, abdominal pain upper, and abdominal discomfort. | ||
| b Fatigue includes fatigue and lethargy. | ||
Adverse Reaction | BLUJEPA N = 309 n (%) | Ceftriaxone and Azithromycin N = 313 n (%) |
Diarrhea | 151 (49) | 30 (10) |
Nausea | 73 (24) | 9 (3) |
Abdominal pain a | 25 (8) | 6 (2) |
Vomiting | 20 (6) | 2 (<1) |
Flatulence | 20 (6) | 1 (<1) |
Dizziness | 16 (5) | 2 (<1) |
Soft feces | 16 (5) | 1 (<1) |
Headache | 10 (3) | 8 (3) |
Fatigue b | 10 (3) | 0 |
Hyperhidrosis | 7 (2) | 0 |
Diarrhea: In Trial 3, diarrhea was reported in 151/309 (49%) uncomplicated urogenital gonorrhea patients receiving BLUJEPA (38% mild severity, 11% moderate severity). Most episodes of diarrhea started on the same day as dosing, with few diarrhea episodes after day 2. The median duration was 2 days. Discontinuation of BLUJEPA due to diarrhea was reported in 1/309 (<1%) uncomplicated urogenital gonorrhea patients.
Adverse Reactions Occurring in Less than 2% of Uncomplicated Urogenital Gonorrhea Patients Receiving BLUJEPA in Trial 3:
Skin and Subcutaneous Tissue: Rash
Cardiac Disorders: Tachycardia
Nervous System Disorders: Dysarthria, presyncope, syncope
Musculoskeletal and Connective Tissue Disorders: Muscle spasms, myalgia, arthralgia
Vascular Disorders: Hot flush
Gastrointestinal Disorders: Hypersalivation, abdominal distension
Eye Disorders: Vision blurred
Ear and Labyrinth Disorders: Vertigo
DRUG INTERACTIONS
- CYP3A4 Inhibitors: Increase gepotidacin exposure. (7.1 )
- Strong CYP3A4 inhibitors: Avoid concomitant use of BLUJEPA with strong CYP3A4 inhibitors. (5.1 , 7.1 )
- Moderate CYP3A4 inhibitors: Avoid concomitant use of BLUJEPA with moderate CYP3A4 inhibitors in patients with uncomplicated urogenital gonorrhea. (5.1 , 7.1 )
- CYP3A4 Inducers: Decrease gepotidacin exposure. (7.1 )
- For uUTI: Avoid concomitant use of BLUJEPA with strong CYP3A4 inducers. (7.1 )
- For uncomplicated urogenital gonorrhea: Avoid concomitant use of BLUJEPA with strong and moderate CYP3A4 inducers. (7.1 )
- CYP3A4 Substrates: Avoid concomitant use of BLUJEPA with drugs that are extensively metabolized by CYP3A4 where minimal concentration changes may lead to serious adverse reactions. (7.2 )
- Digoxin: Due to an increase in digoxin exposures, consider monitoring digoxin serum concentration, as appropriate, with concomitant administration of BLUJEPA. (7.2 )
Effect of Other Drugs on BLUJEPA
CYP3A4 Inhibitors
Indication | Moderate CYP3A4 Inhibitors | Strong CYP3A4 Inhibitors |
Uncomplicated UTI | No dosage adjustment | Avoid concomitant administration of BLUJEPA with strong inhibitors of CYP3A4 due to an increase in gepotidacin exposure [see Warnings and Precautions (5.1 ), Clinical Pharmacology (12.3 )]. |
Uncomplicated Urogenital Gonorrhea | Avoid concomitant administration of BLUJEPA with moderate CYP3A4 inhibitors due to an increase in gepotidacin exposure [see Warnings and Precautions (5.1 ), Clinical Pharmacology (12.3 )]. | Avoid concomitant administration of BLUJEPA with strong inhibitors of CYP3A4 due to an increase in gepotidacin exposure [see Warnings and Precautions (5.1 ), Clinical Pharmacology (12.3 )]. |
CYP3A4 Inducers
Due to decreased gepotidacin exposures, avoid coadministration of BLUJEPA with CYP3A4 inducers as follows:
Effect of BLUJEPA on Other Drugs
CYP3A4 Substrates
Avoid concomitant administration of BLUJEPA with drugs that are extensively metabolized by CYP3A4 where minimal concentration changes may lead to serious adverse reactions [see Clinical Pharmacology (12.3 )] .
Digoxin
Due to an increase in digoxin exposures, consider monitoring digoxin serum concentrations, as appropriate, with concomitant administration of BLUJEPA [see Clinical Pharmacology (12.3 )] .
Cholinergic/Anticholinergic Drugs
As gepotidacin is an acetylcholinesterase inhibitor, there is potential for an exaggerated effect of concomitantly administered succinylcholine‑type neuromuscular blocking agents resulting in a delay in recovery of neuromuscular function. BLUJEPA may augment the effect of other acetylcholinesterase inhibitors. Monitor for exaggerated neuromuscular blockade or excessive cholinergic effects [see Warnings and Precautions (5.2 )] .
There is potential for an antagonistic effect with systemic anticholinergic medications or non‑depolarizing neuromuscular blocking agents. Consider the potential for this interaction if BLUJEPA is administered concomitantly with anticholinergic medications [see Warnings and Precautions (5.2 )] .
Drugs that Prolong the QTc Interval
Due to the increased risk of QTc prolongation, avoid concomitant administration of BLUJEPA with other medications that have the potential to prolong the QTc interval [see Warnings and Precautions (5.1 )] .
DESCRIPTION
BLUJEPA tablets contain gepotidacin mesylate, a triazaacenaphthylene antibacterial that inhibits bacterial DNA gyrase and topoisomerase IV. The chemical name is ( R )-2-((4-(((3,4-dihydro-2 H -pyrano[2,3- c ]pyridin-6-yl)methyl)amino)piperidin-1-yl)methyl)-1,2-dihydro-3 H ,8 H -2a,5,8a-triazaacenaphthylene-3,8-dione methanesulfonate dihydrate. The molecular formula is C 24 H 28 N 6 O 3 ●CH 4 O 3 S●2H 2 O and its molecular mass is 580.66. The structural formula is shown below.
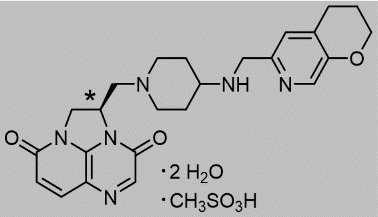
•stereogenic center
Each BLUJEPA oral tablet contains gepotidacin 750 mg (equivalent to 910.7 mg of gepotidacin mesylate [anhydrous]). Inactive ingredients include colloidal silicon dioxide, croscarmellose sodium, magnesium stearate, microcrystalline cellulose, polyethylene glycol, polyvinyl alcohol, talc, titanium dioxide, and yellow iron oxide.
CLINICAL PHARMACOLOGY
Mechanism of Action
BLUJEPA is an antibacterial drug [see Microbiology (12.4 )] .
Pharmacodynamics
The 24-hour free‑drug AUC to minimum inhibitory concentration (MIC) ratio has been shown in animal infection and in vitro pharmacokinetic ‑pharmacodynamic (PK‑PD) models to be the PK‑PD index predictive of gepotidacin antibacterial efficacy.
Cardiac Electrophysiology
The effect of gepotidacin on the QTc interval was evaluated in a randomized, active (moxifloxacin 400 mg) and placebo‑controlled, double‑blind cross‑over trial in healthy subjects who received single intravenous (IV) infusions of gepotidacin over 2 hours. A dose- and concentration‑dependent QTc prolongation effect of gepotidacin was observed. The mean placebo‑corrected change from baseline heart rate values around T max were approximately 6 bpm at 1,000 mg IV (not an approved dosing regimen and route of administration) and approximately 10 bpm at 1,800 mg IV (not an approved dosing regimen and route of administration). The mean placebo‑corrected change from baseline QTcF values around T max were 12 msec at 1,000 mg IV and 22 msec at 1,800 mg IV [see Warnings and Precautions (5.1 )] . The C max of gepotidacin following a single 1,000 mg IV dose (not an approved dosing regimen and route of administration) is approximately 1.2 times that of the C max at steady state for the 1,500 mg oral dose twice daily. The C max of gepotidacin following a single 1,800 mg IV dose (not an approved dosing regimen and route of administration) is approximately 1.2 times that of the C max after the second 3,000 mg oral dose given 12 hours after the first oral dose.
Pharmacokinetics
Pharmacokinetic Parameters
The pharmacokinetic properties of gepotidacin are summarized in Table 4 as mean (standard deviation [SD]) unless otherwise specified.
| a Pharmacokinetic parameters are presented at steady state in patients with uUTI and eGFR greater than or equal to 90 mL/min after oral administration of BLUJEPA 1,500 mg every 12 hours over 5 days, and in simulated results in adults with eGFR greater than or equal to 90 mL/min after oral administration of 2 doses of BLUJEPA (3,000 mg) taken 12 hours apart. | ||
| b AUC 0-12 at steady state in patients with uUTI; simulated AUC 0-24 on day 1 in simulated results with uncomplicated urogenital gonorrhea. | ||
| c Studies evaluating the effect on food were performed with standard and moderate fat meal. Clinical studies were not performed with a high fat meal (1,000 calories, 50% fat). | ||
Dosing Regimen | 1,500 mg Every 12 Hours over 5 Days | 2 Doses of 3,000 mg 12 Hours Apart |
Exposure | ||
C max (mcg/mL) a | 6.3 (1.0) | 11 (2.7) |
AUC (mcg•hour/mL) a,b | 22.8 (4.8) | 75.9 (25.9) |
Dose Proportionality | Approximately dose proportional from 1,500 to 3,000 mg | |
Accumulation | 40% and steady state was achieved by day 3 | |
Absorption | ||
Absolute Bioavailability | ~45% | |
T max (hours) | ~2.0 | |
Effect of food (moderate fat meal) c | No clinically significant effect on PK | |
Distribution | ||
V ss (L) a | 172.9 (42.5) | 188.0 (63.7) |
Plasma Protein Binding | ~25 to ~41% | |
Elimination | ||
Terminal Half-life (hours) a | 9.3 (1.3) | 9.4 (2.3) |
Total Clearance (L/hour) a | 33.4 (6.7) | 34.8 (8.7) |
Metabolism | ||
Primary Pathway | Oxidative metabolism mediated by CYP3A4, producing several circulating metabolites | |
Major Metabolite (%) | M4 which is ~11% of circulating drug‑related materials | |
Excretion | ||
Feces | ~52% (30% unchanged drug) | |
Urine | ~31% (20% unchanged drug; major route of elimination for absorbed gepotidacin) | |
Specific Populations
Modelling and simulation analyses of gepotidacin showed that age, sex, and race have no clinically relevant effect on gepotidacin exposure. Body weight alone does not result in clinically significant effects on gepotidacin exposure [see Use in Specific Populations (8.4 ), (8.5 )] .
Patients with Renal Impairment: The pharmacokinetics of gepotidacin were evaluated in subjects with moderate renal impairment (eGFR 30 to 59 mL/min) and in subjects with severe renal impairment/end stage renal disease (ESRD) on intermittent hemodialysis and not on intermittent hemodialysis (eGFR <30 mL/min). Gepotidacin plasma C max and AUC in subjects with moderate renal impairment were 1.2-fold and 1.5-fold higher than matched healthy controls, respectively. Gepotidacin plasma C max and AUC in severe renal impairment/ESRD not on intermittent hemodialysis were 1.7-fold and 2.1‑fold higher than matched healthy controls, respectively. Gepotidacin plasma C max and AUC in ESRD subjects requiring intermittent hemodialysis were 2.3‑fold and 2.5-fold higher before intermittent hemodialysis than healthy matching subjects, respectively, and were 6.2‑fold and 4.2‑fold higher after intermittent hemodialysis than matched healthy controls, respectively [see Use in Specific Populations (8.6 )] .
Patients with Hepatic Impairment: Mild hepatic impairment did not have a clinically relevant effect on gepotidacin pharmacokinetics. Moderate hepatic impairment resulted in an approximately 1.2‑fold increase in gepotidacin plasma C max and AUC compared with normal hepatic function. In subjects with severe hepatic impairment compared with subjects with normal hepatic function, gepotidacin plasma exposure parameters (C max and AUC) were increased by approximately 1.9‑fold and 1.7‑fold, respectively [see Use in Specific Populations (8.7 )] .
Drug Interaction Studies
Clinical Drug Interaction Studies and Model Informed Approaches:
Effect of CYP3A4 Strong Inhibitors on the Pharmacokinetics of Gepotidacin: Concomitant administration of a strong inhibitor of CYP3A4 (itraconazole; 200 mg per day for 3 days), and a single 1,500 mg dose of BLUJEPA resulted in an increase in the maximum concentration (C max ) of gepotidacin of approximately 1.4‑fold and area under the curve (AUC) of approximately 1.5‑fold [see Drug Interactions (7.1 )] .
Effect of CYP3A4 Strong Inducers on the Pharmacokinetics of Gepotidacin: Concomitant administration of BLUJEPA (single 1,500 mg dose) with a strong CYP3A4 inducer (rifampin; 600 mg once daily for 7 days) resulted in a decrease of 52% in gepotidacin plasma AUC (0-∞) [see Drug Interactions (7.1 )].
Effect of BLUJEPA on Pharmacokinetics of Other Drugs: Concomitant administration of a single 0.5 mg dose of digoxin with two 3,000 mg doses of BLUJEPA (an in vitro P‑glycoprotein inhibitor), given 12 hours apart (not an approved dosage of BLUJEPA), resulted in a 1.5‑fold increase in the digoxin C max (at 3 hours post dose), a 1.1‑fold increase in the digoxin AUC (0-∞) , and a delayed digoxin T max [see Drug Interactions (7.2 )].
Concomitant administration of midazolam (2 mg single dose) with BLUJEPA (2 doses of 3,000 mg, given 12 hours apart; not an approved dosage of BLUJEPA) resulted in a 1.9‑fold increase in midazolam AUC (0-∞) [see Drug Interactions (7.2 )].
Effect of Moderate CYP3A Inhibitors on the Pharmacokinetics of Gepotidacin: Concomitant administration of a moderate CYP3A inhibitor (fluconazole) and single 1,500 mg dose of BLUJEPA is predicted to increase gepotidacin Cmax by 1.3‑fold and AUC by 1.5‑fold [see Drug Interactions (7.1 )].
Effect of Moderate CYP3A Inducers on the Pharmacokinetics of Gepotidacin: Concomitant administration of a moderate CYP3A inducer (efavirenz) and single 1,500 mg dose of BLUJEPA is predicted to decrease AUC and Cmax by 49% and 34%, respectively [see Drug Interactions (7.1 )].
In Vitro Drug Interaction Studies: In vitro, gepotidacin was not an inducer of CYP1A2, 2B6 or 3A4. In vitro, gepotidacin is not a substrate of any of the hepatic organic anion transporting polypeptides (OATPs) 1B1, 1B3, and 2B1, organic anion transporters (OATs) OAT1, OAT2 and OAT3, organic cation transporters (OCTs) OCT2 and OCT3.
In Vitro Studies Where Drug Interaction Potential Was Not Further Evaluated Clinically: In vitro, gepotidacin inhibited multidrug and toxin extrusion (MATEs) MATE1 (IC 50 =16.6 µM), and MATE2‑K (IC 50 =6.9 μM). In vitro, gepotidacin is a substrate of breast cancer resistance protein (max flux rate ratio of 11.0).
Microbiology
Mechanism of Action
BLUJEPA is a triazaacenaphthylene antibacterial that inhibits Type II topoisomerases including bacterial topoisomerase II (DNA gyrase) and topoisomerase IV, thereby inhibiting DNA replication.
Gepotidacin has bactericidal activity against pathogens as determined by time‑kill studies. In vitro studies demonstrated a gepotidacin post‑antibiotic effect ranging from 1.8 to 2.2 hours for E. coli , 1 to >6.6 hours for K. pneumoniae , 1.4 to 3 hours for P. mirabilis , 1 to 2.6 hours for C. freundii , 2.7 to 4.3 hours for S. saprophyticus , 1.2 to 2.7 hours for E. faecalis , and 0.7 to 1 hour for N. gonorrhoeae at 5 times the MIC.
Resistance
Although no clear mechanisms of resistance have been identified for gepotidacin, potential mechanisms that may impact gepotidacin activity are gepotidacin‑specific alterations of DNA gyrase (g yr A, g yr B) and/or topoisomerase IV ( parC , parE ) gene targets, plasmid‑mediated quinolone resistance genes (especially qnr ), and efflux. The following amino acids may be important for gepotidacin activity GyrA P35, V44, D82, A175, GyrB D426, P445 and ParC D79 (based on E. coli numbering) as shown through studies with isogenic mutants in E. coli and K. pneumoniae . The amino acids GyrA D90, A92 and ParC D86 (based on N. gonorrhoeae numbering) as shown through studies with isogenic mutants in N. gonorrhoeae may also be important. A single target‑specific mutation may not significantly impact gepotidacin activity. In studies of certain amino acid substitutions in GyrA and ParC of E.coli and N. gonorrhoeae , a direct relationship between gepotidacin and fluoroquinolone susceptibility was not established. However, in a study of isogenic N. gonorrhoeae strains with mutations in GyrA S91F, A92T, D95G, and ParC D86N, resistance to both gepotidacin and ciprofloxacin was observed. The clinical significance of these findings is unknown. Gepotidacin activity against E. coli , K. pneumoniae , and N. gonorrhoeae is unrelated to beta‑lactam resistance mechanisms.
The frequency of resistance development to gepotidacin due to spontaneous mutations in the gram‑negative pathogens and gram‑positive uropathogens tested in vitro at 10 times MIC ranged from 10 -9 to 10 -10 .
Target‑specific cross‑resistance with other classes of antibacterial drugs has not been identified; therefore, isolates resistant to other drugs may be susceptible to gepotidacin. However, isolates of Enterobacterales with ≥4‑fold increases in gepotidacin MIC have been identified in vitro and in clinical studies. Using an unapproved single dose of ≤3,000 mg of gepotidacin, three microbiological failures were reported among patients from the Phase 2 clinical study with N. gonorrhoeae isolates with a baseline MIC of 1 mcg/mL. Two of these isolates at the end of treatment, had mutations in GyrA and ParC known to be important for gepotidacin binding to the target (ParC D86N, GyrA A92T), and resistance to gepotidacin (MIC increased ≥32‑fold). Using 3,000 mg of gepotidacin followed by a second 3,000 mg dose taken approximately 12 hours later, no N. gonorrhoeae isolates with gepotidacin MIC values >2 mcg/mL were observed, including for pre‑treatment isolates with a ParC D86N mutation.
During clinical studies, gepotidacin demonstrated activity against some isolates of the following multilocus sequence typing (MLST) for E. coli : ST10, ST131, ST1193, ST69, ST95 and ST73. In vitro activity was also demonstrated against the following N. gonorrhoeae MLSTs: ST11422, ST11706, ST1580, ST1583, ST7363, ST7822 and ST9362.
Interaction with Other Antimicrobials
In in vitro studies, no antagonism against Enterobacterales or gram‑positive isolates was observed for gepotidacin in combination with multiple antibacterial drugs, including fluoroquinolones, sulfonamides, cephalosporins, macrolides, tetracyclines, aminoglycosides, glycopeptides, carbapenems, nitrofurans, monobactams, and oxazolidinones, or for N. gonorrhoeae for gepotidacin in combination with fluoroquinolones, cephalosporins, macrolides, tetracyclines, aminoglycosides, pleuromutilins, and aminocyclitols.
Antimicrobial Activity
Gepotidacin has been shown to be active against most isolates of the following microorganisms, both in vitro and in clinical infections [see Indications and Usage (1 )] :
Uncomplicated UTI:
Aerobic bacteria
Gram‑positive bacteria
Enterococcus faecalis
Staphylococcus saprophyticus
Gram ‑ negative bacteria
Citrobacter freundii complex
Escherichia coli
Klebsiella pneumoniae
Uncomplicated Urogenital Gonorrhea:
Aerobic bacteria
Gram ‑ negative bacteria
Neisseria gonorrhoeae
The following in vitro data are available, but their clinical significance is unknown. At least 90 percent of the following bacteria exhibit an in vitro minimum inhibitory concentration (MIC) less than or equal to the susceptible breakpoint for gepotidacin against isolates of similar genus or organism group. However, the efficacy of gepotidacin in treating clinical infections caused by these bacteria has not been established in adequate and well‑controlled clinical trials.
Aerobic bacteria
Gram ‑ negative bacteria
Citrobacter koseri
Klebsiella aerogenes
Klebsiella oxytoca/Raoltella ornithinolytica
Morganella morganii
Proteus mirabilis
Providencia rettgeri
Susceptibility Test Methods
For specific information regarding susceptibility test interpretive criteria and associated test methods and quality control standards recognized by FDA for this drug, please see: https://www.fda.gov/STIC .
NONCLINICAL TOXICOLOGY
Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Long term carcinogenicity studies have not been conducted with gepotidacin.
Mutagenesis
Gepotidacin was positive in an in vitro micronucleus test in human peripheral blood lymphocytes and in an L5178Y mouse lymphoma assay, consistent with the known in vitro clastogenic effects of topoisomerase inhibitors in in vitro mammalian cell assays. Gepotidacin was negative in an in vivo micronucleus test or Comet assay in rat. Based on an overall weight of evidence, gepotidacin is unlikely to be genotoxic.
Impairment of Fertility
In animal studies with gepotidacin, there were no adverse effects on fertility in male and female rats treated for approximately 3 weeks at approximately 2 times the exposure at the MRHD (AUC extrapolated from rats orally administered the same dose for up to 4 weeks). There were no effects on spermatogenesis in rats and dogs at approximately 2 and 3 times the exposure at MRHD, respectively, when treated for up to 13 weeks.
CLINICAL STUDIES
Uncomplicated Urinary Tract Infections
A total of 3,136 female patients with uncomplicated urinary tract infections (uUTI) were randomized in 2 multicenter, parallel‑group, double‑blind, double‑dummy, non‑inferiority (NI) trials (Trial 1 [NCT04020341] and Trial 2 [NCT04187144]). Both trials compared BLUJEPA 1,500 mg (administered orally twice daily with food for 5 days) to nitrofurantoin 100 mg (administered orally twice daily for 5 days).
Patients entered the trials with at least 2 symptoms consistent with uUTI (dysuria, frequency, urgency, or lower abdominal pain) and with evidence of urinary nitrite or pyuria. Patients with any medical condition or presentation suggestive of a complicated UTI, or an upper UTI (e.g., pyelonephritis, urosepsis) were excluded.
Efficacy was assessed as a composite of clinical cure and microbiological response at the Test‑of‑Cure (TOC) Visit (study day 10 to 13) in the microbiological ITT nitrofurantoin‑susceptible (micro‑ITTS) population, which included all patients who received at least 1 dose of study medication, had at least 1 baseline qualifying uropathogen (≥10 5 colony‑forming units [CFU]/mL), and excluded patients with organisms not susceptible to nitrofurantoin. Clinical cure was defined as resolution of all signs and symptoms of acute cystitis present at baseline and no new signs and symptoms without the patient receiving other systemic antimicrobials.
Microbiological response was defined as having all qualifying uropathogens found at baseline at ≥10 5 CFU/mL reduced to <10 3 CFU/mL without the patient receiving other systemic antimicrobials.
Both trials demonstrated non‑inferiority of BLUJEPA to nitrofurantoin for composite response (Table 5 ).
In Trial 1, the micro‑ITTS population consisted of 634 female patients with uUTI (n = 336 BLUJEPA; n = 298 nitrofurantoin). The median age of patients was 54 years, 57% were >50 years of age, 84% were White, 40% had a history of recurrent infection. The U.S. enrolled the greatest percentage of patients (39%). Patient demographic and baseline characteristics were generally balanced between treatment groups [see Adverse Reactions (6.1 )] .
In Trial 2, the micro‑ITTS population consisted of 567 female patients with uUTI (n = 292 BLUJEPA; n = 275 nitrofurantoin). The median age of patients was 51 years, 52% were >50 years of age, 85% were White, 41% had history of recurrent infection. The majority of patients (67%) were enrolled from the United States. Patient demographic and baseline characteristics were generally balanced between treatment groups [see Adverse Reactions (6.1 )] .
Table 5 summarizes the composite response, clinical cure, and microbiological response rates at the TOC visit for Trials 1 and 2 in the micro‑ITTS population.
| micro-ITTS = microbiological Intent to Treat nitrofurantoin-susceptible; CI = confidence interval. | |||
| a BLUJEPA was non-inferior to nitrofurantoin in both studies. The determination of Trial 1 non‑inferiority was based on a planned interim analysis of 607 subjects in micro-ITTS population. The determination of Trial 2 non‑inferiority was based on a planned interim analysis of 541 subjects in micro-ITTS population. | |||
| b Treatment difference (BLUJEPA – nitrofurantoin) calculated using Miettinen and Nurminen Summary Score method adjusting for age group and recurrent/non-recurrent infection status combinations. | |||
Study Endpoint | BLUJEPA n/N (%) | Nitrofurantoin n/N (%) | Treatment Difference (95% CI) b |
Trial 1 | |||
Composite response a | 174/336 (51.8) | 140/298 (47.0) | 5.3 (-2.4, 13.0) |
Clinical cure | 224/336 (66.7) | 196/298 (65.8) | 1.5 (-5.8, 8.8) |
Microbiological response | 244/336 (72.6) | 199/298 (66.8) | 6.0 (-1.2, 13.1) |
Trial 2 | |||
Composite response a | 172/292 (58.9) | 121/275 (44.0) | 14.4 (6.4, 22.4) |
Clinical cure | 199/292 (68.2) | 175/275 (63.6) | 4.3 (-3.4, 12.0) |
Microbiological response | 213/292 (72.9) | 158/275 (57.5) | 15.5 (7.9, 23.1) |
Table 6 summarizes the composite response rates at the TOC Visit for the most common baseline uropathogens across both trials in the micro-ITTS population.
| micro‑ITTS = microbiological Intent to Treat nitrofurantoin-susceptible | ||
| a A patient is counted once under a uropathogen category if multiple qualifying uropathogens within that category are isolated at baseline for the patient. | ||
| b Patients may have been infected with 1 to 2 uropathogens at baseline. | ||
Pathogen b | BLUJEPA a n/N (%) | Nitrofurantoin a n/N (%) |
Escherichia coli | 312/566 (55.1) | 234/520 (45.0) |
Klebsiella pneumoniae | 6/14 (42.9) | 6/16 (37.5) |
Staphylococcus saprophyticus | 9/15 (60.0) | 11/14 (78.6) |
Enterococcus faecalis | 8/14 (57.1) | 2/7 (28.6) |
Citrobacter freundii complex | 8/12 (66.7) | 2/5 (40.0) |
Uncomplicated Urogenital Gonorrhea
A total of 628 patients with suspected uncomplicated urogenital gonorrhea due to Neisseria gonorrhoeae were randomized in an open‑label, active‑controlled, multicenter, multinational trial (Trial 3; NCT04010539). Patients were randomized 1:1 to receive either BLUJEPA (3,000 mg taken orally followed by a second dose of 3,000 mg approximately 12 hours later), or a combination of a single intramuscular 500 mg dose of ceftriaxone and single 1 g oral dose of azithromycin.
Patients were eligible for enrollment if they were ≥12 years old and >45 kg. Patients with confirmed or suspected complicated or disseminated gonorrhea were excluded from the study. The microbiological intent‑to‑treat (micro‑ITT) population, which included patients who had urogenital N. gonorrhoeae isolated at baseline and who were not infected with a strain that was nonsusceptible to ceftriaxone at baseline, consisted of 406 patients (202 for BLUJEPA and 204 for ceftriaxone and azithromycin). The demographic and baseline characteristics in the micro‑ITT population were comparable between treatment groups. In total, 92% were male; 74% White, 15% Black, 6% Asian, 17% Hispanic or Latino; the mean age was 33 years (range: 17 to 64); the mean weight was 76 kg (range: 48 to 120 kg). The primary efficacy endpoint was microbiological success as determined by confirmed bacterial eradication of N. gonorrhoeae at the urogenital body site at the test of cure (TOC) visit (Day 4 to 8) without receipt of other systemic antimicrobials and was assessed in the micro‑ITT population.
Table 7 shows the primary endpoint of the microbiological success rates at the urogenital site at TOC in the micro‑ITT population. Trial 3 demonstrated non‑inferiority of BLUJEPA to the combination of ceftriaxone and azithromycin.
| TOC = Test of Cure; micro‑ITT = microbiological Intent to Treat; CI=confidence interval; n = number of patients in subcategory; N = number of patients in the specified population | |||
| a Calculated using Miettinen and Nurminen Summary Score method adjusting for sex and sexual orientation (female, men who have sex with men, or men who have sex with women) for the BLUJEPA – (ceftriaxone and azithromycin) treatment difference. | |||
| b Unable to determine outcomes were due to missing data, culture processed beyond 24 hours, TOC out of window, or use of another systemic antimicrobial prior to the TOC visit | |||
BLUJEPA n/N (%) | Ceftriaxone and Azithromycin n/N (%) | Treatment Difference (95% CI) a | |
Microbiological success | 187/202 (92.6) | 186/204 (91.2) | -0.1 (-5.6, 5.5) |
Microbiological failure | 15/202 (7.4) | 18/204 (8.8) | |
Bacterial persistence by culture | 0 | 0 | |
Unable to determine b | 15/202 (7.4) | 18/204 (8.8) | |
HOW SUPPLIED/STORAGE AND HANDLING
BLUJEPA tablets are supplied as yellow, film-coated, capsule-shaped tablets debossed with “GS GU3” on one side and plain on the other side, containing 750 mg of gepotidacin.
Bottle of 8 tablets (NDC 0173-0922-38).
Bottle of 20 tablets (NDC 0173-0922-45).
Store at 20°C to 25°C (68°F to 77°F); excursions permitted between 15°C and 30°C (59°F and 86°F). [See USP Controlled Room Temperature].
Mechanism of Action
BLUJEPA is an antibacterial drug [see Microbiology (12.4 )] .