Emtricitabine - Emtricitabine capsule prescribing information
WARNING: POSTTREATMENT ACUTE EXACERBATION OF HEPATITIS B
Severe acute exacerbations of hepatitis B (HBV) have been reported in patients who are coinfected with HIV-1 and HBV and have discontinued emtricitabine. Hepatic function should be monitored closely with both clinical and laboratory follow-up for at least several months in patients who are coinfected with HIV-1 and HBV and discontinue emtricitabine. If appropriate, initiation of anti-hepatitis B therapy may be warranted [see Warnings and Precautions (5.1) ] .
INDICATIONS AND USAGE
Emtricitabine capsules are indicated in combination with other antiretroviral agents for the treatment of HIV-1 infection.
DOSAGE AND ADMINISTRATION
- Testing: Prior to or when initiating emtricitabine capsules test for hepatitis B virus infection. (2.1 )
- Emtricitabine capsules may be taken without regard to food. (2.2 )
- Adult Patients (18 years of age and older) (2.3 ):
- One 200 mg capsule administered once daily orally.
- Pediatric Patients (3 months through 17 years of age) (2.5 ):
- For children weighing more than 33 kg who can swallow an intact capsule, one 200 mg capsule administered once daily orally.
- Dose interval adjustment in adult patients with renal impairment (2.6 ):
| Formulation | Creatinine Clearance (mL/min) | |||
| ≥50 mL/min | 30 to 49 mL/min | 15 to 29 mL/min | <15 mL/min or on hemodialysis a | |
| Capsule (200 mg) | 200 mg every 24 hours | 200 mg every 48 hours | 200 mg every 72 hours | 200 mg every 96 hours |
a. Hemodialysis Patients: If dosing on day of dialysis, give dose after dialysis.
Testing Prior to Initiation of Treatment with Emtricitabine Capsules
Prior to or when initiating emtricitabine capsules, test patients for hepatitis B virus infection [see Warnings and Precautions (5.1) ].
Recommended Dosage
Emtricitabine capsules are taken by mouth once daily and may be taken without regard to food [see Clinical Pharmacology (12.3) ].
Recommended Dosage in Adult Patients (18 years of age and older)
One 200 mg capsule administered once daily orally.
Recommended Dosage in Pediatric Patients (3 months through 17 years of age)
For pediatric patients weighing more than 33 kg who can swallow an intact capsule, one 200 mg capsule administered once daily orally.
Dosage Adjustment in Patients with Renal Impairment
Table 1 provides dosage interval adjustment for patients with renal impairment. No dosage adjustment is necessary for patients with mild renal impairment (creatinine clearance 50 to 80 mL/min). The safety and effectiveness of dose adjustment recommendations in patients with moderate to severe renal impairment (creatinine clearance below 50 mL/min) have not been clinically evaluated. Therefore, clinical response to treatment and renal function should be closely monitored in these patients [see Warnings and Precautions (5.4) , Use in Specific Populations (8.6) ].
| a. Hemodialysis Patients: If dosing on day of dialysis, give dose after dialysis. | ||||
| Formulation | Creatinine Clearance (mL/min) | |||
| ≥50 mL/min | 30 to 49 mL/min | 15 to 29 mL/min | <15 mL/min or on hemodialysis a | |
| Capsule (200 mg) | 200 mg every 24 hours | 200 mg every 48 hours | 200 mg every 72 hours | 200 mg every 96 hours |
There are insufficient data available to make dosage recommendations in pediatric patients with renal impairment.
DOSAGE FORMS AND STRENGTHS
Emtricitabine Capsules, 200 mg are cream cap/cream body, size ‘1’ hard gelatin capsule filled with white to off-white granular powder and imprinted with ‘F’ on cap and ‘36’ on body with black ink.
USE IN SPECIFIC POPULATIONS
Pregnancy
Pregnancy Exposure Registry
There is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to emtricitabine during pregnancy. Healthcare providers are encouraged to register patients by calling the Antiretroviral Pregnancy Registry (APR) at 1-800-258-4263.
Risk Summary
Available data from the APR show no increase in the overall risk of major birth defects with first trimester exposure for emtricitabine (FTC) (2.3%) compared with the background rate for major birth defects of 2.7% in a U.S. reference population of the Metropolitan Atlanta Congenital Defects Program (MACDP) (see Data) . The rate of miscarriage for individual drugs is not reported in the APR. In the U.S. general population, the estimated background risk of miscarriage in clinically recognized pregnancies is 15 to 20%.
In animal reproduction studies, no adverse developmental effects were observed when FTC was administered at exposures ≥60 times that of the recommended daily dose of emtricitabine (see Data) .
Data
Human Data
Based on prospective reports to the APR of exposures to FTC-containing regimens during pregnancy resulting in live births (including over 2,700 exposed in the first trimester and over 1,200 exposed in the second/third trimester), there was no increase between FTC and overall birth defects compared with the background birth defect rate of 2.7% in a U.S. reference population of the MACDP. The prevalence of birth defects in live births was 2.4% (95% CI: 1.9% to 3.1%) with first trimester exposure to FTC-containing regimens and 2.3% (95% CI: 1.5% to 3.3%) with the second/third trimester exposure to FTC-containing regimens.
Prospective reports from the APR of overall major birth defects in pregnancies exposed to FTC are compared with a U.S. background major birth defect rate. Methodologic limitations of the APR include the use of MACDP as the external comparator group. Limitations of using an external comparator include differences in methodology and populations, as well as confounding due to the underlying disease.
Additionally, published observational studies on FTC exposure in pregnancy have not shown an increased risk for major malformations.
Animal Data
FTC was administered orally to pregnant mice (at 0, 250, 500, or 1,000 mg/kg/day), and rabbits (at 0, 100, 300, or 1,000 mg/kg/day) through organogenesis (on gestation days 6 through 15, and 7 through 19, respectively). No significant toxicological effects were observed in embryo-fetal toxicity studies performed with FTC in mice at exposures (AUC) approximately 60 times higher and in rabbits at approximately 120 times higher than human exposures at the recommended daily dose. In a pre/postnatal development study in mice, FTC was administered orally at doses up to 1,000 mg/kg/day; no significant adverse effects directly related to drug were observed in the offspring exposed daily from before birth ( in utero ) through sexual maturity at daily exposures (AUC) of approximately 60 times higher than human exposures at the recommended daily dose.
Lactation
Risk Summary
The Centers for Disease Control and Prevention recommend that HIV-1 infected mothers not breastfeed their infants to avoid risking postnatal transmission of HIV-1.
Based on published data, FTC has been shown to be present in human breast milk. It is not known if FTC affects milk production or has effects on the breastfed child.
Because of the potential for: (1) HIV transmission (in HIV-negative infants); (2) developing viral resistance (in HIV-positive infants); and (3) adverse reactions in a breastfed infant similar to those seen in adults, instruct mothers not to breastfeed if they are taking emtricitabine.
Pediatric Use
The safety and efficacy of FTC in patients between 3 months and 21 years of age is supported by data from three open-label, nonrandomized clinical trials in which FTC was administered to 169 HIV-1 infected treatment-naïve and experienced (defined as virologically suppressed on a 3TC containing regimen for which FTC was substituted for 3TC) subjects [see Clinical Studies (14.4) ] .
The pharmacokinetics of FTC were studied in 20 neonates born to HIV-1 positive mothers [see Clinical Studies (14.4) ] . All neonates were HIV-1 negative at the end of the trial; the efficacy of FTC in preventing or treating HIV-1 could not be determined.
Geriatric Use
Clinical trials of emtricitabine did not include sufficient numbers of subjects aged 65 years and over to determine whether they respond differently from younger subjects. In general, dose selection for the elderly patient should be cautious, keeping in mind the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
Renal Impairment
Modify the dose or dosing interval for emtricitabine in patients with creatinine clearance below 50 mL/min or in patients with end stage renal disease requiring dialysis [see Dosage and Administration (2.6) ] .
CONTRAINDICATIONS
Emtricitabine capsules are contraindicated in patients with previously demonstrated hypersensitivity to any of the components of the products.
WARNINGS AND PRECAUTIONS
Severe Acute Exacerbation of Hepatitis B in Patients Coinfected with HIV-1 and HBV
All patients should be tested for the presence of chronic Hepatitis B virus (HBV) before or when initiating emtricitabine [see Dosage and Administration (2.1) ]. Severe acute exacerbations of hepatitis B (e.g., liver decompensation and liver failure) have been reported in patients who are coinfected with HIV-1 and HBV and have discontinued emtricitabine. Patients who are coinfected with HIV-1 and HBV who discontinue emtricitabine should be closely monitored with both clinical and laboratory follow-up for at least several months after stopping treatment. If appropriate, initiation of anti-hepatitis B therapy may be warranted, especially in patients with advanced liver disease or cirrhosis, since posttreatment exacerbation of hepatitis may lead to hepatic decompensation and liver failure.
5.2 Immune Reconstitution Syndrome
Immune reconstitution syndrome has been reported in patients treated with combination antiretroviral therapy, including emtricitabine. During the initial phase of combination antiretroviral treatment, patients whose immune system responds may develop an inflammatory response to indolent or residual opportunistic infections (such as Mycobacterium avium infection, cytomegalovirus, Pneumocystis jirovecii pneumonia [PCP], or tuberculosis), which may necessitate further evaluation and treatment.
Autoimmune disorders (such as Graves' disease, polymyositis, and Guillain-Barré syndrome) have also been reported to occur in the setting of immune reconstitution; however, the time to onset is more variable, and can occur many months after initiation of treatment.
5.3 Lactic Acidosis/Severe Hepatomegaly with Steatosis
Lactic acidosis and severe hepatomegaly with steatosis, including fatal cases, have been reported with the use of nucleoside analogs, including FTC, alone or in combination with other antiretrovirals. Treatment with emtricitabine should be suspended in any patient who develops clinical or laboratory findings suggestive of lactic acidosis or pronounced hepatotoxicity (which may include hepatomegaly and steatosis even in the absence of marked transaminase elevations).
Dose Adjustment in Patients with New Onset or Worsening Renal Impairment
Emtricitabine is principally eliminated by the kidney. Reduction of the dosage of emtricitabine is recommended for patients with impaired renal function [see Dosage and Administration (2.6) , Use in Specific Populations (8.6) , and Clinical Pharmacology (12.3) ] .
ADVERSE REACTIONS
The following adverse reactions are discussed in other sections of the labeling:
- Severe Acute Exacerbation of Hepatitis B in Patients Coinfected with HIV-1 and HBV [see Warnings and Precautions (5.1) ] .
- Immune Reconstitution Syndrome [see Warnings and Precautions (5.2) ] .
- Lactic Acidosis/Severe Hepatomegaly with Steatosis [see Warnings and Precautions (5.3) ].
Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Adverse Reactions from Clinical Trials Experience in Adults
More than 2,000 adult subjects with HIV-1 infection have been treated with emtricitabine alone or in combination with other antiretroviral agents for periods of 10 days to 200 weeks in clinical trials.
The most common adverse reactions (incidence greater than or equal to 10%, any severity) identified from any of the three large, controlled clinical trials include headache, diarrhea, nausea, fatigue, dizziness, depression, insomnia, abnormal dreams, rash, abdominal pain, asthenia, increased cough, and rhinitis.
In Trials 301A and 303, the most common adverse reactions that occurred in subjects receiving emtricitabine with other antiretroviral agents were headache, diarrhea, nausea, and rash, which were generally mild to moderate. Approximately 1% of subjects discontinued participation in the clinical trials due to these events. All adverse reactions were reported with similar frequency in emtricitabine and control treatment groups except for skin discoloration, which was reported with higher frequency in the emtricitabine-treated group.
Skin discoloration, manifested by hyperpigmentation on the palms or soles, was generally mild and asymptomatic. The mechanism and clinical significance are unknown.
A summary of emtricitabine treatment-emergent clinical adverse reactions in Trials 301A and 303 is provided in Table 2.
| AZT=zidovudine; d4T=stavudine; NNRTI/PI=non-nucleoside reverse transcriptase inhibitor/protease inhibitor; 3TC=lamivudine; EFV=efavirenz. a. Rash event includes rash, pruritus, maculopapular rash, urticaria, vesiculobullous rash, pustular rash, and allergic reaction. | ||||
| 303 | 301A | |||
| Emtricitabine + AZT/d4T + NNRTI/PI (N=294) | 3TC + AZT/d4T + NNRTI/PI (N=146) | Emtricitabine + didanosine + EFV (N=286) | d4 T + didanosine + EFV (N=285) | |
| Body as a Whole | ||||
| Asthenia | 16% | 10% | 12% | 17% |
| Headache | 13% | 6% | 22% | 25% |
| Abdominal pain | 8% | 11% | 14% | 17% |
| Digestive System | ||||
| Diarrhea | 23% | 18% | 23% | 32% |
| Nausea | 18% | 12% | 13% | 23% |
| Vomiting | 9% | 7% | 9% | 12% |
| Dyspepsia | 4% | 5% | 8% | 12% |
| Musculoskeletal | ||||
| Myalgia | 4% | 4% | 6% | 3% |
| Arthralgia | 3% | 4% | 5% | 6% |
| Nervous System | ||||
| Insomnia | 7% | 3% | 16% | 21% |
| Depressive disorders | 6% | 10% | 9% | 13% |
| Paresthesia | 5% | 7% | 6% | 12% |
| Dizziness | 4% | 5% | 25% | 26% |
| Neuropathy/peripheral neuritis | 4% | 3% | 4% | 13% |
| Abnormal dreams | 2% | <1% | 11% | 19% |
| Respiratory | ||||
| Rhinitis | 18% | 12% | 12% | 10% |
| Increased cough | 14% | 11% | 14% | 8% |
| Skin | ||||
| Rash event a | 17% | 14% | 30% | 33% |
Laboratory Abnormalities : Laboratory abnormalities in these trials occurred with similar frequency in the emtricitabine and comparator groups. A summary of Grades 3 to 4 laboratory abnormalities is provided in Table 3.
| a. ULN = Upper limit of normal | ||||
| 303 | 301A | |||
| Emtricitabine + AZT/d4T + NNRTI/PI (N=294) | 3TC + AZT/d4T + NNRTI/PI (N=146) | Emtricitabine + Didanosine + EFV (N=286) | d4 T + Didanosine + EFV (N=285) | |
| Any ≥ Grade 3 Laboratory Abnormality | 31% | 28% | 34% | 38% |
| ALT (>5.0 × ULN a ) | 2% | 1% | 5% | 6% |
| AST (>5.0 × ULN) | 3% | <1% | 6% | 9% |
| Bilirubin (>2.5 × ULN) | 1% | 2% | <1% | <1% |
| Creatine kinase (>4.0 × ULN) | 11% | 14% | 12% | 11% |
| Neutrophils (<750 mm 3 ) | 5% | 3% | 5% | 7% |
| Pancreatic amylase (>2.0 × ULN) | 2% | 2% | <1% | 1% |
| Serum amylase (>2.0 × ULN) | 2% | 2% | 5% | 10% |
| Serum glucose <40 or >250 mg/dL) | 3% | 3% | 2% | 3% |
| Serum lipase (>2.0 × ULN) | <1% | <1% | 1% | 2% |
| Triglycerides (>750 mg/dL) | 10% | 8% | 9% | 6% |
In Trial 934, 511 antiretroviral-naïve subjects received efavirenz (EFV) administered in combination with either emtricitabine + tenofovir disoproxil fumarate (TDF) (N=257) or AZT/3TC (N=254) for 144 weeks. The most common adverse reactions (incidence greater than or equal to 10%, all grades) included diarrhea, nausea, fatigue, headache, dizziness, depression, insomnia, abnormal dreams, and rash. Table 4 provides the treatment-emergent adverse reactions (Grades 2 to 4) occurring in greater than or equal to 5% of subjects treated in any treatment group.
| a. Frequencies of adverse reactions are based on all treatment-emergent adverse events, regardless of relationship to study drug. b. From Weeks 96 to 144 of the trial, subjects received TRUVADA ® with EFV in place of emtricitabine + TDF with EFV. c. Rash event includes rash, exfoliative rash, rash generalized, rash macular, rash maculo-papular, rash pruritic, and rash vesicular. | ||
| Emtricitabine + TDF + EFV b | AZT/3TC + EFV | |
| N=257 | N=254 | |
| Fatigue | 9% | 8% |
| Depression | 9% | 7% |
| Nausea | 9% | 7% |
| Diarrhea | 9% | 5% |
| Dizziness | 8% | 7% |
| Upper respiratory tract infections | 8% | 5% |
| Sinusitis | 8% | 4% |
| Rash event c | 7% | 9% |
| Headache | 6% | 5% |
| Insomnia | 5% | 7% |
| Nasopharyngitis | 5% | 3% |
| Vomiting | 2% | 5% |
Laboratory Abnormalities: Laboratory abnormalities observed in Trial 934 were generally consistent with those seen in previous trials (Table 5).
| a. From Weeks 96 to 144 of the trial, subjects received TRUVADA with EFV in place of emtricitabine + TDF with EFV. | ||
| Emtricitabine + TDF + EFV a | AZT/3TC + EFV | |
| N=257 | N=254 | |
| Any ≥ Grade 3 Laboratory Abnormality | 30% | 26% |
| Fasting Cholesterol (>240 mg/dL) | 22% | 24% |
| Creatine Kinase (M: >990 U/L) (F: >845 U/L) | 9% | 7% |
| Serum Amylase (>175 U/L) | 8% | 4% |
| Alkaline Phosphatase (>550 U/L) | 1% | 0% |
| AST (M: >180 U/L) (F: >170 U/L) | 3% | 3% |
| ALT (M: >215 U/L) (F: >170 U/L) | 2% | 3% |
| Hemoglobin (<8.0 mg/dL) | 0% | 4% |
| Hyperglycemia (>250 mg/dL) | 2% | 1% |
| Hematuria (>75 RBC/HPF) | 3% | 2% |
| Glycosuria (3+) | <1% | 1% |
| Neutrophils (<750/mm 3 ) | 3% | 5% |
| Fasting Triglycerides (>750 mg/dL) | 4% | 2% |
Adverse Reactions from Clinical Trials Experience in Pediatric Subjects
Assessment of adverse reactions in pediatric subjects is based on data from Trial 203, an open label, uncontrolled trial of 116 HIV-1 infected subjects who received FTC through 48 weeks. The adverse reaction profile in pediatric subjects was generally comparable to that observed in clinical trials of emtricitabine in adult subjects [see Adverse Reactions (6.1) ] . Hyperpigmentation was more frequent in children. Additional adverse reactions identified from this trial include anemia.
Selected treatment-emergent adverse events, regardless of causality, reported in subjects during 48 weeks of treatment were the following: infection (44%), hyperpigmentation (32%), increased cough (28%), vomiting (23%), otitis media (23%), rash (21%), rhinitis (20%), diarrhea (20%), fever (18%), pneumonia (15%), gastroenteritis (11%), abdominal pain (10%), and anemia (7%). Treatment-emergent Grades 3 to 4 laboratory abnormalities were experienced by 9% of pediatric subjects, including elevated amylase (>2.0 x ULN) (n=4), decreased neutrophils (<750/mm 3 ) (n=3), elevated ALT (>5 x ULN) (n=2), elevated CPK (>4 x ULN) (n=2) and one subject each with elevated bilirubin (>3.0 x ULN), elevated GGT (>10 x ULN), elevated lipase (>2.5 x ULN), decreased hemoglobin (<7 g/dL), and decreased glucose (<40 mg/dL).
DRUG INTERACTIONS
The potential for drug interactions with emtricitabine has been studied in combination with AZT, indinavir, d4T, famciclovir, and tenofovir DF (TDF). There were no clinically significant drug interactions for any of these drugs. Drug interactions trials are described elsewhere in the labeling [see Clinical Pharmacology (12.3) ].
DESCRIPTION
Emtricitabine (FTC) is a synthetic nucleoside analog with activity against human immunodeficiency virus type 1 (HIV-1) reverse transcriptase.
The chemical name of FTC is 5-fluoro-1-(2 R ,5 S )-[2-(hydroxymethyl)-1,3-oxathiolan-5-yl]cytosine. Emtricitabine is the (-) enantiomer of a thio analog of cytidine, which differs from other cytidine analogs in that it has a fluorine in the 5-position.
It has a molecular formula of C 8 H 10 FN 3 O 3 S and a molecular weight of 247.24. It has the following structural formula:
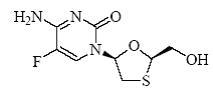
Emtricitabine is a white to off-white powder with a solubility of approximately 112 mg/mL in water at 25°C. The partition coefficient (log P) for FTC is −0.43 and the pKa is 2.65.
Emtricitabine capsules are for oral administration. Each capsule contains 200 mg of FTC and the inactive ingredients: crospovidone, gelatin, magnesium stearate, microcrystalline cellulose, povidone, sodium lauryl sulphate, titanium dioxide and yellow iron oxide. The capsules are imprinted with edible ink containing black iron oxide, potassium hydroxide, propylene glycol, shellac and strong ammonia solution.
CLINICAL PHARMACOLOGY
Mechanism of Action
Emtricitabine is an antiretroviral drug [see Microbiology (12.4) ] .
Pharmacokinetics
Adults
The pharmacokinetic properties of FTC were evaluated in healthy subjects and HIV-1-infected subjects. Emtricitabine pharmacokinetics are similar between these populations.
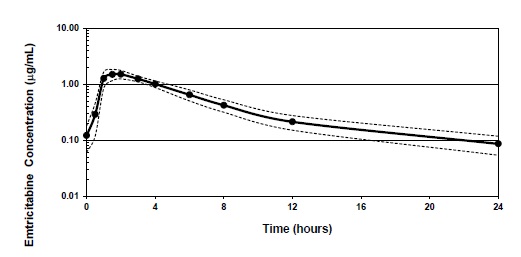
Figure 1 shows the mean steady-state plasma FTC concentration-time profile in 20 HIV‑1-infected subjects receiving emtricitabine capsules.
Figure 1 Mean (± 95% CI) Steady-State Plasma FTC Concentrations in HIV‑1 Infected Adults (N=20)
Absorption
Emtricitabine is rapidly and extensively absorbed following oral administration, with peak plasma concentrations occurring at 1 to 2 hours postdose. Following multiple dose oral administration of emtricitabine capsules to 20 HIV-1 infected subjects, the (mean ± SD) steady-state plasma FTC peak concentration (C max ) was 1.8 ± 0.7 mcg/mL and the area-under the plasma concentration-time curve over a 24-hour dosing interval (AUC) was 10.0 ± 3.1 mcg·hr/mL. The mean steady-state plasma trough concentration at 24 hours postdose was 0.09 mcg/mL. The mean absolute bioavailability of emtricitabine capsules was 93%, while the mean absolute bioavailability of emtricitabine oral solution was 75%. The relative bioavailability of emtricitabine oral solution was approximately 80% of emtricitabine capsules.
The multiple dose pharmacokinetics of FTC are dose proportional over a dose range of 25 to 200 mg.
Distribution
In vitro binding of FTC to human plasma proteins was less than 4% and independent of concentration over the range of 0.02 to 200 mcg/mL. At peak plasma concentration, the mean plasma to blood drug concentration ratio was ~1.0 and the mean semen to plasma drug concentration ratio was ~4.0.
Metabolism
Following administration of radiolabelled FTC, complete recovery of the dose was achieved in urine (~86%) and feces (~14%). Thirteen percent (13%) of the dose was recovered in urine as three putative metabolites. The biotransformation of FTC includes oxidation of the thiol moiety to form the 3’-sulfoxide diastereomers (~9% of dose) and conjugation with glucuronic acid to form 2’-O-glucuronide (~4% of dose). No other metabolites were identifiable.
Elimination
The plasma FTC half-life is approximately 10 hours. The renal clearance of FTC is greater than the estimated creatinine clearance, suggesting elimination by both glomerular filtration and active tubular secretion. There may be competition for elimination with other compounds that are also renally eliminated.
Effects of Food on Oral Absorption
Emtricitabine capsules and oral solution may be taken with or without food. Emtricitabine systemic exposure (AUC) was unaffected while C max decreased by 29% when emtricitabine capsules were administered with food (an approximately 1000 kcal high-fat meal). Emtricitabine systemic exposure (AUC) and C max were unaffected when 200 mg emtricitabine oral solution was administered with either a high-fat or low-fat meal.
Specific Populations
Geriatric Patients
The pharmacokinetics of FTC have not been fully evaluated in the elderly (65 years of age and older).
Pediatric Patients
The pharmacokinetics of FTC at steady state were determined in 77 HIV-1 infected pediatric subjects, and the pharmacokinetic profile was characterized in four age groups (Table 6). The FTC exposure achieved in pediatric subjects receiving a daily dose of 6 mg/kg up to a maximum of 240 mg oral solution or a 200 mg capsule is similar to exposures achieved in adult subjects receiving a once-daily dose of 200 mg.
The pharmacokinetics of FTC were studied in 20 neonates born to HIV-1 positive mothers. Each mother received prenatal and intrapartum combination antiretroviral therapy. Neonates received up to 6 weeks of AZT prophylactically after birth. The neonates were administered two short courses of FTC oral solution (each 3 mg/kg once daily × 4 days) during the first 3 months of life. The AUC observed in neonates who received a daily dose of 3 mg/kg of FTC was similar to the AUC observed in pediatric subjects aged 3 months to 17 years who received a daily dose of FTC as a 6 mg/kg oral solution up to 240 mg or as a 200 mg capsule (Table 6).
| a. Two pharmacokinetic evaluations were conducted in 20 neonates over the first 3 months of life. Median (range) age of infant on day of pharmacokinetic evaluation was 26 (5 to 81) days. b. Mean (range). | |||||
| HIV-1- exposed Neonates | HIV-1 Infected Pediatric Subjects | ||||
| Age | 0 to 3 mo (N=20) a | 3 to 24 mo (N=14) | 25 mo to 6 yr (N=19) | 7 to 12 yr (N=17) | 13 to 17 yr (N=27) |
| Formulation | 0 | 0 | 0 | 10 | 26 |
| Capsule (n) | |||||
| Oral Solution (n) | 20 | 14 | 19 | 7 | 1 |
| Dose (mg/kg) b | 3.1 (2.9 to 3.4) | 6.1 (5.5 to 6.8) | 6.1 (5.6 to 6.7) | 5.6 (3.1 to 6.6) | 4.4 (1.8 to 7.0) |
| C max (mcg/mL) | 1.6 ± 0.6 | 1.9 ± 0.6 | 1.9 ± 0.7 | 2.7 ± 0.8 | 2.7 ± 0.9 |
| AUC (mcg · hr/mL) | 11.0 ± 4.2 | 8.7 ± 3.2 | 9.0 ± 3.0 | 12.6 ± 3.5 | 12.6 ± 5.4 |
| T 1/2 (hr) | 12.1 ± 3.1 | 8.9 ± 3.2 | 11.3 ± 6.4 | 8.2 ± 3.2 | 8.9 ± 3.3 |
Gender
FTC pharmacokinetics are similar in adult male and female subjects.
Race
No pharmacokinetic differences due to race have been identified.
Patients with Renal Impairment
The pharmacokinetics of FTC are altered in subjects with renal impairment [see Warnings and Precautions (5.4) ] . In adult subjects with creatinine clearance below 50 mL/min or with end-stage renal disease (ESRD) requiring dialysis, C max and AUC of FTC were increased (Table 7). The effects of renal impairment on FTC pharmacokinetics in pediatric patients are not known.
| Creatinine Clearance (mL/min) | >80 (N=6) | 50 to 80 (N=6) | 30 to 49 (N=6) | <30 (N=5) | ESRD a <30 (N=5) |
|---|---|---|---|---|---|
| Baseline creatinine clearance (mL/min) | 107 ± 21 | 59.8 ± 6.5 | 40.9 ± 5.1 | 22.9 ± 5.3 | 8.8 ± 1.4 |
| C max (mcg/mL) | 2.2 ± 0.6 | 3.8 ± 0.9 | 3.2 ± 0.6 | 2.8 ± 0.7 | 2.8 ± 0.5 |
| AUC (mcg.hr/mL) | 11.8 ± 2.9 | 19.9 ± 1.2 | 25.1 ± 5.7 | 33.7± 2.1 | 53.2 ± 9.9 |
| CL/F (mL/min) | 302 ± 94 | 168 ± 10 | 138 ± 28 | 99 ± 6 | 64 ± 12 |
| CLr (mL/min) | 213 ± 89 | 121 ± 39 | 69 ± 32 | 30 ± 11 | NA b |
a. ESRD subjects requiring dialysis
b. NA = Not Applicable
Patients with Hepatic Impairment
The pharmacokinetics of FTC have not been studied in subjects with hepatic impairment; however, FTC is not significantly metabolized by liver enzymes, so the impact of liver impairment should be limited.
Assessment of Drug Interactions
At concentrations up to 14-fold higher than those observed in vivo , FTC did not inhibit in vitro drug metabolism mediated by any of the following human CYP isoforms: CYP1A2, CYP2A6, CYP2B6, CYP2C9, CYP2C19, CYP2D6, and CYP3A4. FTC did not inhibit the enzyme responsible for glucuronidation (uridine-5’-disphosphoglucuronyl transferase). Based on the results of these in vitro experiments and the known elimination pathways of FTC, the potential for CYP-mediated interactions involving FTC with other medicinal products is low.
Emtricitabine has been evaluated in healthy volunteers in combination with TDF, AZT, indinavir, famciclovir, and d4T. Tables 8 and 9 summarize the pharmacokinetic effects of coadministered drug on FTC pharmacokinetics and effects of FTC on the pharmacokinetics of coadministered drug.
| a. All interaction trials conducted in healthy volunteers. b. ↑ = Increase; ⇔ = No Effect; NA = Not Applicable | ||||||
| Coadministered Drug | Dose of Coadministered Drug (mg) | FTC Dose (mg) | N | % Change of FTC Pharmacokinetic Parameters b (90% CI) | ||
| C max | AUC | C min | ||||
| Famciclovir | 500 × 1 | 200 × 1 | 12 | ⇔ | ⇔ | NA |
| Indinavir | 800 × 1 | 200 × 1 | 12 | ⇔ | ⇔ | NA |
| Stavudine | 40 × 1 | 200 × 1 | 6 | ⇔ | ⇔ | NA |
| Tenofovir DF | 300 once daily × 7 days | 200 once daily × 7 days | 17 | ⇔ | ⇔ | ↑ 20 (↑ 12 to ↑ 29) |
| Zidovudine | 300 twice daily × 7 days | 200 once daily × 7 days | 27 | ⇔ | ⇔ | ⇔ |
| a. All interaction trials conducted in healthy volunteers. b. ↑ = Increase; ⇔ = No Effect; NA = Not Applicable | ||||||
| Coadministered Drug | Dose of Coadministered Drug (mg) | FTC Dose (mg) | N | % Change of Coadministered Drug Pharmacokinetic Parameters b (90% CI) | ||
| C max | AUC | C min | ||||
| Famciclovir | 500 × 1 | 200 × 1 | 12 | ⇔ | ⇔ | NA |
| Indinavir | 800 × 1 | 200 × 1 | 12 | ⇔ | ⇔ | NA |
| Stavudine | 40 × 1 | 200 × 1 | 6 | ⇔ | ⇔ | NA |
| Tenofovir DF | 300 once daily × 7 days | 200 once daily × 7 days | 17 | ⇔ | ⇔ | ⇔ |
| Zidovudine | 300 twice daily × 7 days | 200 once daily × 7 days | 27 | ↑ 17 (↑ 0 to ↑ 38) | ↑ 13 (↑ 5 to ↑ 20) | ⇔ |
Microbiology
Mechanism of Action
FTC, a synthetic nucleoside analog of cytidine, is phosphorylated by cellular enzymes to form emtricitabine 5'-triphosphate (FTC-TP), which inhibits the activity of the HIV-1 reverse transcriptase (RT) by competing with the natural substrate deoxycytidine 5'‑triphosphate and by being incorporated into nascent viral DNA resulting in chain termination. FTC-TP is a weak inhibitor of mammalian DNA polymerases α, β, ε, and mitochondrial DNA polymerase γ.
Antiviral Activity
The antiviral activity of FTC against laboratory and clinical isolates of HIV-1 was assessed in lymphoblastoid cell lines, the MAGI-CCR5 cell line, and peripheral blood mononuclear cells. The 50% effective concentration (EC 50 ) values for FTC were in the range of 0.0013 to 0.64 µM (0.0003 to 0.158 mcg/mL). The median EC 50 , range, and number of isolates for the laboratory isolates were 0.07, 0.009 to 0.62, 10; 0.011, 0.002 to 0.03, 12; 0.055, 0.0015 to 0.09, 4 respectively for lymphoblastoid, PBMC, and reporter cells. The median EC 50 , range, and number of isolates for the clinical isolates were 0.05, 0.0105 to 0.64, 15; 0.015, 0.004 to 0.028, 6 respectively for lymphoblastoid cells and PBMCs. No antagonism was observed in drug combination studies of emtricitabine with nucleoside reverse transcriptase inhibitors (NRTIs) (abacavir, lamivudine, stavudine, tenofovir, zidovudine), non-nucleoside reverse transcriptase inhibitors (NNRTIs) (delavirdine, efavirenz, nevirapine), and protease inhibitors (amprenavir, nelfinavir, ritonavir, saquinavir). FTC displayed antiviral activity in cell culture against HIV-1 clades A, B, C, D, E, F, and G (EC 50 values ranged from 0.007 to 0.075 µM) and showed strain-specific activity against HIV-2 (EC 50 values ranged from 0.007 to 1.5 µM in PBMCs and MAGI cells).
Resistance
FTC-resistant isolates of HIV-1 have been selected in cell culture and in vivo . Genotypic analysis of these isolates showed that the reduced susceptibility to FTC was associated with a valine or isoleucine (M184V/I) substitution in the HIV-1 RT.
FTC-resistant isolates of HIV-1 have been recovered from some subjects treated with FTC alone or in combination with other antiretroviral agents. In a clinical trial of treatment-naïve subjects treated with emtricitabine, didanosine, and efavirenz (EFV) [see Clinical Studies (14.2)] , viral isolates from 37.5% of subjects with virologic failure showed reduced susceptibility to FTC. Genotypic analysis of these isolates showed that the resistance was due to M184V/I substitutions in the HIV-1 RT.
In a clinical trial of treatment-naïve subjects treated with either emtricitabine, TDF, and EFV or AZT/3TC and EFV [see Clinical Studies (14.2) ] , resistance analysis was performed on HIV-1 isolates from all confirmed virologic failure subjects with greater than 400 copies/mL of HIV-1 RNA at Week 144 or early discontinuation. Development of EFV resistance-associated substitutions occurred most frequently and was similar between the treatment arms. The M184V amino acid substitution, associated with resistance to emtricitabine and 3TC, was observed in 2/19 analyzed subject isolates in the emtricitabine + TDF group and in 10/29 analyzed subject isolates in the AZT/3TC group. Through 144 weeks of Trial 934, no subjects have developed a detectable K65R substitution in their HIV-1 as analyzed through standard genotypic analysis.
Cross Resistance
Cross-resistance among certain NRTIs has been recognized. FTC-resistant isolates (M184V/I) were cross-resistant to 3TC but retained susceptibility in cell culture to AZT, didanosine, d4T, and tenofovir, and to NNRTIs (delavirdine, EFV, and nevirapine). HIV-1 isolates containing the K65R substitution, selected in vivo by abacavir, didanosine, and tenofovir, demonstrated reduced susceptibility to inhibition by FTC. Viruses harboring substitutions conferring reduced susceptibility to AZT and d4T (M41L, D67N, K70R, L210W, T215Y/F, K219Q/E) or didanosine (L74V) remained sensitive to FTC. HIV-1 containing the K103N substitution associated with resistance to NNRTIs was susceptible to FTC.
NONCLINICAL TOXICOLOGY
Carcinogenesis, Mutagenesis, Impairment of Fertility
In long-term oral carcinogenicity studies of FTC, no drug-related increases in tumor incidence were found in mice at doses up to 750 mg/kg/day (26 times the human systemic exposure at the therapeutic dose of 200 mg/day) or in rats at doses up to 600 mg/kg/day (31 times the human systemic exposure at the therapeutic dose).
Emtricitabine was not genotoxic in the reverse mutation bacterial test (Ames test) or mouse lymphoma or mouse micronucleus assays.
Emtricitabine did not affect fertility in male rats at approximately 140-fold or in male and female mice at approximately 60-fold higher exposures (AUC) than in humans given the recommended 200 mg daily dose. Fertility was normal in the offspring of mice exposed daily from before birth ( in utero ) through sexual maturity at daily exposures (AUC) of approximately 60-fold higher than human exposures at the recommended 200 mg daily dose.
CLINICAL STUDIES
Overview of Clinical Trials
The efficacy and safety of emtricitabine were evaluated in the trials summarized in Table 10.
Table 10 Trials Conducted with Emtricitabine in Adult and Pediatric Subjects
| Trial | Population | Trial Arms (N) a | Timepoint (Weeks) |
| Trial 934 b (NCT00112047) | HIV-1 treatment-naïve adults | Emtricitabine+TDF+EFV (227) AZT/3TC+EFV (229) | 144 |
| Trial 301A c (NCT00006208) | Emtricitabine+didanosine+EFV (286) d4T+didanosine+EFV (285) | 48 | |
| Trial 303 b (NCT00002416) | HIV-1 treatment- experienced adults | Emtricitabine+AZT/d4T+NNRTI/PI (294) 3TC+AZT/d4T+NNRTI/PI (146) | 48 |
| Trial 202 d (NCT00016718) | HIV-1 treatment-naïve and experienced pediatrics | Emtricitabine+didanosine+EFV (43) | 48 |
| Trial 203 d (NCT00743340) | Emtricitabine+d4T+lopinavir/ritonavir (116) | 48 | |
| Trial 211 d (NCT00642291) | Emtricitabine+didanosine+EFV (16) | 48 |
a. Randomized and dosed.
b. Randomized, open-label, active-controlled trial.
c. Randomized, double-blind, active-controlled trial.
d. Nonrandomized, open-label trial.
Clinical Trial Results in Treatment-Naïve Adults
Trial 934
Data through 144 weeks are reported for Trial 934, a randomized, open-label, active-controlled multicenter clinical trial comparing emtricitabine + TDF administered in combination with EFV versus AZT/3TC fixed-dose combination administered in combination with EFV in 511 antiretroviral-naïve subjects. From Weeks 96 to 144 of the trial, subjects received FTC/TDF fixed-dose combination with EFV in place of emtricitabine + TDF with EFV. Subjects had a mean age of 38 years (range 18 to 80); 86% were male, 59% were Caucasian, and 23% were Black. The mean baseline CD4+ cell count was 245 cells/mm 3 (range 2 to 1191) and median baseline plasma HIV-1 RNA was 5.01 log 10 copies/mL (range 3.56 to 6.54). Subjects were stratified by baseline CD4+ cell count (< or ≥200 cells/mm 3 ); 41% had CD4+ cell counts <200 cells/mm 3 and 51% of subjects had baseline viral loads >100,000 copies/mL. Table 11 provides treatment outcomes through 48 and 144 weeks for those subjects who did not have EFV resistance at baseline.
Table 11 Virologic Outcomes of Randomized Treatment at Week 48 and 144 (Trial 934)
| Outcomes | Week 48 | Week 144 | ||
| Emtricitabine +TDF +EFV (N=244) | AZT/3TC +EFV (N=243) | Emtricitabine +TDF +EFV (N=227) a | AZT/3TC +EFV (N=229) a | |
| Responder b | 84% | 73% | 71% | 58% |
| Virologic failure c | 2% | 4% | 3% | 6% |
| Rebound | 1% | 3% | 2% | 5% |
| Never suppressed | 0% | 0% | 0% | 0% |
| Change in antiretroviral regimen | 1% | 1% | 1% | 1% |
| Death | <1% | 1% | 1% | 1% |
| Discontinued due to adverse event | 4% | 9% | 5% | 12% |
| Discontinued for other reasons d | 10% | 14% | 20% | 22% |
a. Subjects who were responders at Week 48 or Week 96 (HIV-1 RNA <400 copies/mL) but did not consent to continue in the trial after Week 48 or Week 96 were excluded from analysis.
b. Subjects achieved and maintained confirmed HIV-1 RNA <400 copies/mL through Weeks 48 and 144.
c. Includes confirmed viral rebound and failure to achieve confirmed <400 copies/mL through Weeks 48 and 144.
d. Includes lost to follow-up, subject withdrawal, noncompliance, protocol violation and other reasons.
Through Week 48, 84%, and 73% of subjects in the emtricitabine + TDF group and the AZT/3TC group, respectively, achieved and maintained HIV-1 RNA <400 copies/mL (71% and 58% through Week 144). The difference in the proportion of subjects who achieved and maintained HIV-1 RNA <400 copies/mL through 48 weeks largely results from the higher number of discontinuations due to adverse events and other reasons in the AZT/3TC group in this open-label trial. In addition, 80% and 70% of subjects in the emtricitabine + TDF group and the AZT/3TC group, respectively, achieved and maintained HIV-1 RNA <50 copies/mL through Week 48 (64% and 56% through Week 144). The mean increase from baseline in CD4+ cell count was 190 cells/mm 3 in the emtricitabine + TDF group and 158 cells/mm 3 in the AZT/3TC group at Week 48 (312 and 271 cells/mm 3 at Week 144).
Through 48 weeks, 7 subjects in the emtricitabine + TDF group and 5 subjects in the AZT/3TC group experienced a new CDC Class C event (10 and 6 subjects through 144 weeks).
Trial 301A
Trial 301A was a 48-week double-blind, active-controlled, multicenter clinical trial comparing emtricitabine (200 mg once daily) administered in combination with didanosine and EFV versus d4T, didanosine, and EFV in 571 antiretroviral naïve adult subjects. Subjects had a mean age of 36 years (range 18 to 69); 85% were male, 52% Caucasian, 16% African-American, and 26% Hispanic. Subjects had a mean baseline CD4+ cell count of 318 cells/mm 3 (range 5 to 1317) and a median baseline plasma HIV-1 RNA of 4.9 log 10 copies/mL (range 2.6 to 7.0). Thirty-eight percent of subjects had baseline viral loads >100,000 copies/mL and 31% had CD4+ cell counts <200 cells/mL. Table 12 provides treatment outcomes through 48 weeks.
Table 12 Virologic Outcomes of Randomized Treatment at Week 48 (Trial 301A)
| Outcomes | Emtricitabine + Didanosine + EFV (N=286) | d4T + Didanosine + EFV (N=285) |
| Responder a | 81% (78%) | 68% (59%) |
| Virologic Failure b | 3% | 11% |
| Death | 0% | <1% |
| Discontinuation Due to Adverse Event | 7% | 13% |
| Discontinuation for Other Reasons c | 9% | 8% |
a. Subjects achieved and maintained confirmed HIV RNA <400 copies/mL (<50 copies/mL) through Week 48. b. Includes subjects who failed to achieve virologic suppression or rebounded after achieving virologic suppression.
c. Includes lost to follow-up, subject withdrawal, non-compliance, protocol violation, and other reasons.
The mean increase from baseline in CD4+ cell count was 168 cells/mm 3 for the emtricitabine arm and 134 cells/mm 3 for the d4T arm.
Through 48 weeks, 5 subjects (1.7%) in the emtricitabine group experienced a new CDC Class C event compared to 7 subjects (2.5%) in the d4T group.
Clinical Trial Results in Treatment-Experienced Adults
Trial 303
Trial 303 was a 48-week, open-label, active-controlled, multicenter clinical trial comparing emtricitabine (200 mg once daily) to 3TC, in combination with d4T or AZT and a protease inhibitor or NNRTI in 440 adult subjects who were on a 3TC-containing triple-antiretroviral drug regimen for at least 12 weeks prior to trial entry and had HIV-1 RNA ≤400 copies/mL.
Subjects were randomized 1:2 to continue therapy with 3TC (150 mg twice daily) or to switch to emtricitabine (200 mg once daily). All subjects were maintained on their stable background regimen. Subjects had a mean age of 42 years (range 22 to 80); 86% were male, 64% Caucasian, 21% African-American, and 13% Hispanic. Subjects had a mean baseline CD4+ cell count of 527 cells/mm 3 (range 37 to 1909), and a median baseline plasma HIV-1 RNA of 1.7 log 10 copies/mL (range 1.7 to 4.0).
The median duration of prior antiretroviral therapy was 27.6 months. Table 13 provides treatment outcomes through 48 weeks.
Table 13 Virologic Outcomes of Randomized Treatment at Week 48 (Trial 303)
| Outcomes | Emtricitabine + AZT/d4T + NNRTI/PI (N=294) | 3TC + AZT/d4T + NNRTI/PI (N=146) |
| Responder a | 77% (67%) | 82% (72%) |
| Virologic Failure b | 7% | 8% |
| Death | 0% | <1% |
| Discontinuation Due to Adverse Event | 4% | 0% |
| Discontinuation for Other Reasons c | 12% | 10% |
a. Subjects achieved and maintained confirmed HIV RNA <400 copies/mL (<50 copies/mL) through Week 48.
b. Includes subjects who failed to achieve virologic suppression or rebounded after achieving virologic suppression.
c. Includes lost to follow-up, subject withdrawal, non-compliance, protocol violation, and other reasons.
The mean increase from baseline in CD4+ cell count was 29 cells/mm 3 for the emtricitabine arm and 61 cells/mm 3 for the 3TC arm.
Through 48 weeks, in the emtricitabine group 2 subjects (0.7%) experienced a new CDC Class C event compared to 2 subjects (1.4%) in the 3TC group.
Clinical Trial Results in Pediatrics
In three open-label, nonrandomized clinical trials, FTC was administered to 169 HIV-1 infected treatment-naïve and experienced (defined as virologically suppressed on a 3TC-containing regimen for which FTC was substituted for 3TC) subjects between 3 months and 21 years of age. Subjects received once-daily emtricitabine oral solution (6 mg/kg to a maximum of 240 mg/day) or emtricitabine capsules (a single 200 mg capsule once daily) in combination with at least two other antiretroviral agents.
Subjects had a mean age of 7.9 years (range 0.3 to 21); 49% were male, 15% Caucasian, 61% Black, and 24% Hispanic. Subjects had a median baseline HIV-1 RNA of 4.6 log 10 copies/mL (range 1.7 to 6.4) and a mean baseline CD4+ cell count of 745 cells/mm 3 (range 2 to 2650). Through 48 weeks of therapy, the overall proportion of subjects who achieved and sustained an HIV-1 RNA <400 copies/mL was 86%, and <50 copies/mL was 73%. The mean increase from baseline in CD4+ cell count was 232 cells/mm 3 (-945, +1512). The adverse reaction profile observed during these clinical trials was similar to that of adult subjects, with the exception of the occurrence of anemia and higher frequency of hyperpigmentation in children [see Adverse Reactions (6.1) ] .
The pharmacokinetics of FTC were studied in 20 neonates born to HIV-1 positive mothers. Each mother received prenatal and intrapartum combination antiretroviral therapy. Neonates received up to 6 weeks of AZT prophylactically after birth. The neonates were administered two short courses of FTC oral solution (each 3 mg/kg once daily × 4 days) during the first 3 months of life. FTC exposures in neonates were similar to the exposures achieved in subjects aged 3 months to 17 years [see Clinical Pharmacology (12.3) ] . During the two short dosing periods on FTC, there were no safety issues identified in the treated neonates. All neonates were HIV-1 negative at the end of the trial; the efficacy of FTC in preventing or treating HIV‑1 could not be determined.
HOW SUPPLIED/STORAGE AND HANDLING
Emtricitabine Capsules, 200 mg are cream cap/cream body, size ‘1’ hard gelatin capsule filled with white to off-white granular powder and imprinted with ‘F’ on cap and ‘36’ on body with black ink.
Bottles of 30 NDC 65862-301-30
Bottles of 500 NDC 65862-301-05
- Store at 20° to 25°C (68° to 77°F); excursions permitted to 15° to 30°C (59° to 86°F) [see USP Controlled Room Temperature].
- Keep container tightly closed.
Mechanism of Action
Emtricitabine is an antiretroviral drug [see Microbiology (12.4) ] .