Get your patient on Forzinity - Elamipretide Hydrochloride injection (Elamipretide Hydrochloride)
Forzinity - Elamipretide Hydrochloride injection prescribing information
INDICATIONS AND USAGE
FORZINITY is indicated to improve muscle strength in adult and pediatric patients with Barth syndrome weighing at least 30 kg.
This indication is approved under accelerated approval based on an improvement in knee extensor muscle strength, an intermediate clinical endpoint [see Clinical Studies (14) ] . Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial.
DOSAGE AND ADMINISTRATION
For patients weighing 30 kg and greater, the recommended dosage is 40 mg subcutaneously once daily. (2.1 )
Reduce the dose in adults with severe renal impairment. (2.2 , 8.6 ).
Recommended Dosage
The recommended dosage of FORZINITY in patients weighing at least 30 kg is 40 mg subcutaneously once daily.
Patients should receive the dose at the same time each day.
Missed Dose
If a dose is missed, skip the dose and take the next dose of FORZINITY at the scheduled time. Do not take a double dose of FORZINITY.
Dosage Modifications For Renal Impairment
Refer to Table 1 for dosage modifications in adults with renal impairment.
| Estimated Glomerular Filtration Rate (eGFR) (mL/minute) | Recommended Dosage |
|---|---|
| Greater than or equal to 30 mL/minute | 40 mg subcutaneously once daily |
| Less than 30 mL/minute and NOT on dialysis | 20 mg subcutaneously once daily |
There is insufficient information to recommend a dosage regimen in adults with eGFR less than 30 mL/minute and on dialysis.
There is insufficient information to recommend a dosage regimen in pediatric patients weighing 30 kg or greater with renal impairment.
Important Administration Instructions
FORZINITY is a clear, colorless to yellow aqueous solution that contains the preservative, benzyl alcohol. Visually inspect each vial of FORZINITY for particulate matter and cloudiness prior to administration, whenever solution and container permit. Do not use if the solution is cloudy or particulate matter is present.
Adult patients or caregivers may administer FORZINITY after proper training in preparing FORZINITY vials for administration, if a healthcare provider determines that it is appropriate, and with medical follow-up as necessary.
Use aseptic technique to prepare and administer FORZINITY. Administer FORZINITY by subcutaneous injection in the abdomen (at least 2 inches from the navel) or outer thigh and rotate the injection site daily. Do not inject where the skin is tender, bruised, red, or hard. Avoid injecting into scars or stretch marks. Refer to the Instructions for Use for preparation and administration.
FORZINITY is for single-patient-use only.
Do not mix other products in the same syringe.
Discard vials 8 days after first opening.
DOSAGE FORMS AND STRENGTHS
Injection: 280 mg/3.5 mL (80 mg/mL), sterile, clear, colorless to yellow aqueous solution for injection supplied as single-patient-use vials.
USE IN SPECIFIC POPULATIONS
Pregnancy
Risk Summary
Barth Syndrome is a rare, X-linked, recessive, genetic disorder and is not likely to affect females. Therefore, there are no data with FORZINITY use in pregnant women to evaluate for a drug-related risk of major birth defects, miscarriage, or other adverse maternal or fetal outcomes. In animal reproduction studies, no adverse developmental outcomes occurred at any dose tested (see Data ). FORZINITY contains benzyl alcohol as a preservative. Because benzyl alcohol is rapidly metabolized by a pregnant woman, benzyl alcohol exposure in the fetus is unlikely.
The background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Data
Animal Data
In pregnant rats, once-daily intravenous infusion of elamipretide during the period of organogenesis [gestation day (GD) 7 to GD 17] did not result in embryo-fetal developmental toxicity at doses tested up to 10 mg/kg/day, approximately 4 times the clinical exposure at the maximum recommended human dose (MRHD) of 40 mg/day, based on area under the concentration-time curve (AUC).
In pregnant rabbits, intravenous infusion with elamipretide once daily during the period of organogenesis (GD 7 to GD 19) did not result in embryo-fetal developmental toxicity at doses tested up to 50 mg/kg/day, approximately 10 times the clinical exposure at the MRHD, based on AUC.
In a pre- and postnatal study in rats, elamipretide was subcutaneously administered at doses of 0, 5, 10 or 15 mg/kg/day throughout pregnancy and lactation (GD 6 to Lactation Day 20). No adverse developmental effects were observed at doses up to 15 mg/kg/day, approximately 6-times the clinical exposure at the MRHD, based on AUC.
Lactation
Risk Summary
Barth Syndrome is a rare, X-linked, recessive, genetic disorder and is not likely to affect females. Therefore, there is no data to evaluate the presence of FORZINITY in human milk, the effect on the breastfed infant, or the effects on milk production.
FORZINITY contains benzyl alcohol. Because benzyl alcohol is rapidly metabolized by a lactating female, benzyl alcohol exposure in the breastfed infant is unlikely. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for FORZINITY and any potential adverse effects on the breastfed infant from FORZINITY or from the underlying maternal condition.
Pediatric Use
The safety and effectiveness of FORZINITY to improve muscle strength have been established in pediatric patients with Barth syndrome weighing at least 30 kg. Use of FORZINITY for this indication is supported by improvement in knee extensor muscle strength, an intermediate clinical endpoint, observed in an open-label extension study of FORZINITY that included seven pediatric patients aged 12 years and older. [see Dosage and Administration (2.1) , and Clinical Studies (14) ] .
The safety and effectiveness of FORZINITY have not been established in pediatric patients weighing less than 30 kg.
FORZINITY is not approved for use in neonates. Serious adverse reactions, including fatal reactions, of new onset or worsening metabolic acidosis that progressed to neurotoxicity, and in some cases gasping syndrome, have been reported in low-birth weight neonates and preterm neonates who received BA containing drugs intravenously (FORZINITY is not approved for intravenous use) [see Dosage and Administration (2.1) ] . Gasping syndrome is a life-threatening condition in neonates caused by BA toxicity that is characterized by new onset or worsening metabolic acidosis with gradual neurological deterioration, seizures, intracranial hemorrhage, hematologic abnormalities, skin breakdown, hepatic and renal failure, hypotension, bradycardia, and gasping respirations followed by death. In reported cases, BA in amounts of 99 to 234 mg/kg/day produced blood BA levels of 6.6 to 14.9 mg/dL, but the minimum amount of BA at which gasping syndrome may occur in neonates is not known (FORZINITY contains 20 mg of BA per mL) [see Warnings and Precautions (5.1) ] .
Geriatric Use
Clinical studies of FORZINITY did not enroll subjects with Barth syndrome aged 65 years and older.
Renal Impairment
No dosage adjustment is required for patients with mild (eGFR 60 to 89 mL/min) or moderate (eGFR 30 to 59 mL/min) renal impairment. The FORZINITY dose should be reduced by one half administered subcutaneously once daily in adults with severe renal impairment (eGFR less than 30 mL/min) who are not on dialysis [see Dosage and Administration (2.2) ] . FORZINITY has not been studied in patients with renal failure on dialysis [see Clinical Pharmacology (12.3) ] .
CONTRAINDICATIONS
Serious hypersensitivity to elamipretide or any of the excipients in FORZINITY [see Warnings and Precautions (5.2) ].
WARNINGS AND PRECAUTIONS
- Benzyl alcohol toxicity : Do not use in neonates. (5.1 )
Risk of Benzyl Alcohol Toxicity in Neonates
FORZINITY is not approved for use in neonates. Serious adverse reactions, including fatal reactions, of new onset or worsening metabolic acidosis that progressed to neurotoxicity, and in some cases gasping syndrome, have been reported in low-birth weight neonates (less than 2,500 grams) and preterm neonates (gestational age less than 34 weeks) who received benzyl alcohol (BA)-containing drugs intravenously. FORZINITY is not approved for intravenous use [see Dosage and Administration (2.1) ] . Gasping syndrome is a life-threatening condition in neonates caused by BA toxicity and is primarily characterized by multiorgan dysfunction secondary to metabolic acidosis, which leads to gasping respirations and death. The minimum amount of BA at which these serious adverse reactions, including fatal reactions, may occur is not known (FORZINITY contains 20 mg of BA per mL). [See Use in Specific Populations (8.4) .]
Hypersensitivity
Hypersensitivity reactions, including serious allergic reactions requiring emergency medical intervention, have been reported in patients receiving FORZINITY. These reactions have included skin manifestations such as rash, papular lesions, and eczematous dermatitis, as well as respiratory symptoms including cough [see Adverse Reactions (6.1) ] . Reactions may occur within minutes to months after treatment initiation.
Monitor patients for signs and symptoms of hypersensitivity reactions during treatment. If a serious hypersensitivity reaction occurs, do not administer further doses of FORZINITY and institute appropriate emergency treatment, including epinephrine, antihistamines, and corticosteroids as clinically indicated. Patients who have experienced a serious hypersensitivity reaction should not be rechallenged with FORZINITY [see Contraindications (4) ] .
For mild to moderate skin reactions, consider treatment with topical corticosteroids and oral antihistamines. Consider discontinuation of FORZINITY if persistent or severe skin reactions develop.
ADVERSE REACTIONS
Most common adverse reactions are injection site reactions. (6.1 )
To report SUSPECTED ADVERSE REACTIONS, contact Stealth BioTherapeutics Inc. at 1-844-444-6486 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
In the FORZINITY clinical development program, 12 male patients aged 12 to 35 years with genetically-confirmed Barth syndrome received treatment with daily subcutaneous injections of 40 mg FORZINITY. Eleven of these 12 patients were Caucasian.
Patients first participated in a double-blind, placebo-controlled crossover trial where they were randomized to one of two sequences:
- 12 weeks of FORZINITY in Period 1 then a 4-week washout followed by 12 weeks of placebo in Period 2 or
- 12 weeks of placebo in Period 1 then a 4-week washout followed by 12 weeks of FORZINITY in Period 2
Ten patients completed the randomized trial and entered the open-label extension period where they received FORZINITY once daily. Eight of these patients received FORZINITY for 168 weeks, three of whom received FORZINITY for a total of 192 weeks.
Adverse reactions occurring more commonly on FORZINITY than on placebo include injection site reactions such as injection site erythema, pain, induration, pruritus, bruising, and urticaria (Table 2).
| Combined | ||
|---|---|---|
| (Periods 1 and 2) | ||
| Elamipretide | Placebo | |
| N=12 | N=12 | |
| n (%) | n (%) | |
| Any local administration reaction | 12 (100) | 8 (67) |
| Injection site erythema | 12 (100) | 3 (25) |
| Injection site induration | 8 (67) | 2 (17) |
| Injection site pruritus | 8 (67) | 2 (17) |
| Injection site pain | 9 (75) | 5 (42) |
| Injection site bruising | 3 (25) | 0 |
| Injection site urticaria | 3 (25) | 0 |
| Injection site hemorrhage | 0 | 1 (8) |
Eosinophilia
Increases in absolute eosinophil counts were noted frequently in studies where duration of administration of FORZINITY was 30 days or greater. Eosinophil counts generally peaked around 90 days after initial exposure (mean increase from baseline ~0.5 to 0.6 × 10 3 /uL) and returned to baseline levels after 6 to 12 months of continuous exposure or after discontinuation of FORZINITY. The elevation in eosinophils was not associated with clinical manifestations or changes in other laboratory parameters.
DESCRIPTION
FORZINITY contains elamipretide, a mitochondrial cardiolipin binder. Elamipretide is isolated as a hydrochloride salt that is freely soluble in water. The chemical name for elamipretide hydrochloride is L-Phenylalaninamide, D-arginyl-2,6-dimethyl-L-tyrosyl-L-lysyl-, hydrochloride (1:3). Its molecular formula is C 32 H 49 N 9 O 5 ∙3HCl and its molecular weight is 749.2.
The structure of elamipretide hydrochloride is:
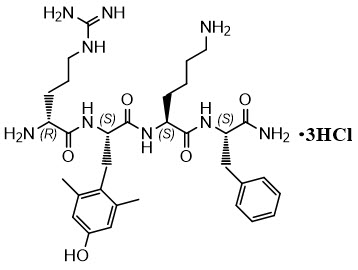
All amino acid residues in FORZINITY have the L configuration except for arginine which has the D configuration. The peptide sequence is denoted as D-Arg-2,6-dimethyl-Tyr-Lys-Phe-NH 2 .
FORZINITY is a ready-to-use sterile, clear, colorless to yellow aqueous solution supplied as single-patient-use vials containing 3.5 mL solution for subcutaneous injection. Each 0.5 mL dose of FORZINITY contains 40 mg of elamipretide (equivalent to 46.8 mg elamipretide hydrochloride), 10 mg benzyl alcohol as a preservative, and 2.07 mg monobasic sodium phosphate (as monohydrate). The product may contain hydrochloric acid or sodium hydroxide to adjust pH. The pH of FORZINITY solution is 4.7 to 6.1.
CLINICAL PHARMACOLOGY
Mechanism of Action
FORZINITY is a mitochondrial cardiolipin binder that localizes to the inner mitochondrial membrane and improves mitochondrial morphology and function.
Pharmacodynamics
Effects on QTc Interval
Cardiac electrophysiology
Clinically significant QTc interval prolongation was not observed at 3 times the peak concentration of the maximum recommended FORZINITY dose.
Pharmacokinetics
Elamipretide exposure increases proportionally over a dose range of 2 to 80 mg following daily subcutaneous injections with minimal accumulation.
Absorption
Maximum elamipretide concentrations were reached between 0.5 to 1 hour after subcutaneous administration. The absolute bioavailability following subcutaneous administration is approximately 92%. FORZINITY exposure is comparable after subcutaneous injection to the thigh or to the abdomen.
Distribution
Elamipretide is distributed throughout total body water with an approximate volume of distribution of 0.5 L/kg. There is low binding to plasma proteins (approximately 39%).
Elimination
Metabolism
Elamipretide is metabolized via sequential C-terminal degradation to the M1 tripeptide and M2 dipeptide metabolites, which do not have pharmacological activity.
Excretion
Elamipretide and its metabolites M1 and M2 are excreted in the urine. At 48 hours post-dose, approximately 100% of the FORZINITY dose was recovered in the urine as either elamipretide, M1, or M2 in patients with normal renal function.
Specific Populations
Patients with Renal Impairment
Elamipretide exposure (AUC) increased by 39% in subjects with creatinine clearance 60 to 89 mL/min, 75% in subjects with creatinine clearance 30 to 59 mL/min, and 125% in subjects with creatinine clearance less than 30 mL/min not on dialysis. Renal impairment was categorized based on 24-hour measured urinary creatinine clearance. There was minimal accumulation of elamipretide with daily dosing, regardless of the severity of renal impairment. There was a significant increase in exposure of the M1 and M2 metabolites, up to 280% and 640%, respectively, in subjects with severe renal impairment not on dialysis (creatinine clearance less than 30 mL/min).
While the effect of renal impairment on elamipretide pharmacokinetics was characterized using 24-hour measured urinary creatinine clearance, analyses conducted with estimated glomerular filtration (CKD-EPI equation) support the recommendations for use in this specific population [see Dosage and Administration (2.2) and Use in Specific Populations (8.6) ] .
Patients with Hepatic Impairment
No hepatic metabolism was observed for elamipretide in vitro. Hepatic impairment is not expected to alter the pharmacokinetics (PK) of elamipretide.
Drug Interaction Studies
In Vitro Studies
CYP enzymes: Elamipretide does not inhibit CYP1A, CYP2D6, CYP2E1, CYP2B6, CYP2C8, CYP2C9, CYP2C19, or CYP3A. Elamipretide does not induce metabolism by CYP1A2, CYP2B6, or CYP3A4.
Drug transporters: Elamipretide does not inhibit the activity of OCT2, BCRP, OAT1, OAT3, OATP1B1, OATP1B3, P-gp, or MATE2-K. Elamipretide is an inhibitor of MATE1 (IC 50 3.53 μM).
In Vivo Clinical Studies
Aspirin: administration of elamipretide (0.1 mg/kg/hour for 4 hours via intravenous infusion) starting 4 hours after a single dose of 650 mg aspirin did not affect the PK of aspirin and its metabolite, salicylic acid. There was no impact on the antiplatelet activity of aspirin when co-administered with elamipretide.
Clopidogrel: administration of elamipretide (0.1 mg/kg/hour for 4 hours via intravenous infusion) starting 4 hours after a single dose of 300 mg clopidogrel did not significantly affect the PK of clopidogrel. There was no impact on the anti-thrombotic activity of clopidogrel when co-administered with elamipretide.
Unfractionated heparin: administration of elamipretide (0.25 mg/kg/hour for 4 hours via intravenous infusion) had no significant impact on activated partial thromboplastin time (aPTT) and anti-factor Xa activity of unfractionated heparin, when co-administered 7 hours after starting the unfractionated heparin infusion.
NONCLINICAL TOXICOLOGY
Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies have not been conducted with elamipretide.
Elamipretide was negative in an in vitro bacterial reverse mutation assay, a chromosomal aberration assay in Chinese hamster ovary cells, and an in vivo rat bone marrow micronucleus assay.
Subcutaneous doses of elamipretide in rats of up to 20 mg/kg/day, approximately 5-times the clinical exposure at the MRHD of 40 mg, did not adversely affect fertility or reproductive performance.
CLINICAL STUDIES
FORZINITY was evaluated in a randomized, double-blind, placebo-controlled, crossover trial and its 192-week, open-label, single-arm extension period.
The randomized trial evaluated the efficacy and safety of once daily FORZINITY 40 mg injected subcutaneously for 12 weeks in 12 subjects ≥12-years-old and >30 kg with genetically confirmed Barth syndrome. The primary endpoints for the randomized trial were distance walked during 6-minute walk test and Total Fatigue Score on the Barth syndrome Symptom Assessment. FORZINITY was not superior to placebo on these primary endpoints. Ten subjects completed the randomized trial and entered the extension period designed to evaluate long-term safety and tolerability of FORZINITY. Eight of these 10 subjects participated through Week 168 of the extension period.
Knee extensor muscle strength measured by handheld dynamometry was evaluated as one of the secondary endpoints in the randomized trial and in the extension period. Increases in knee extensor muscle strength were not observed during the randomized trial but were observed during the extension period. At the pre-dose baseline visit at the start of the randomized trial, median (min, max) muscle strength was 124 (92, 176) newtons. Table 3 shows descriptive changes from pre-dose baseline for knee extensor muscle strength during the randomized controlled trial and extension period.
| Visit | N | Median Change | Min, Max Change | |
|---|---|---|---|---|
| Randomized Controlled Trial | Week 12 Placebo | 12 | -5 | -31, 48 |
| Week 12 Elamipretide | 12 | 4 | -41, 86 | |
| Open-Label Extension Period | Week 12 | 10 | 34 | 7, 95 |
| Week 24 | 9 | 68 | 3, 90 | |
| Week 36 | 8 | 57 | 9, 92 | |
| Week 48 | 8 | 41 | -2, 144 | |
| Week 72 | 8 | 35 | -4, 100 | |
| Week 168 | 8 | 63 | 38, 78 |
HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
FORZINITY (elamipretide) injection is a sterile, clear, colorless to yellow aqueous solution supplied as:
- Carton containing four 280 mg/3.5 mL (80 mg/mL) single-patient-use vials (NDC 72507-800-04)
Storage
Store refrigerated at 2°C to 8°C (36°F to 46°F). Do not freeze. Following the first dose, the opened vial can be stored either refrigerated 2°C to 8°C (36°F to 46°F) or at room temperature between 20°C to 25°C (68°F to 77°F). Discard vials 8 days after first opening.
Mechanism of Action
FORZINITY is a mitochondrial cardiolipin binder that localizes to the inner mitochondrial membrane and improves mitochondrial morphology and function.