Get your patient on Fosphenytoin Sodium - Fosphenytoin Sodium injection (Fosphenytoin Sodium)
Fosphenytoin Sodium - Fosphenytoin Sodium injection prescribing information
INDICATIONS AND USAGE
Fosphenytoin sodium injection, USP is indicated for the control of generalized tonic-clonic status epilepticus and prevention and treatment of seizures occurring during neurosurgery. Fosphenytoin can also be substituted, short-term, for oral phenytoin. Fosphenytoin should be used only when oral phenytoin administration is not possible. Fosphenytoin must not be given orally.
DOSAGE AND ADMINISTRATION
The dose, concentration, and infusion rate of intravenous fosphenytoin should always be expressed as phenytoin sodium equivalents (PE). There is no need to perform molecular weight-based adjustments when converting between fosphenytoin and phenytoin sodium doses. Fosphenytoin should always be prescribed and dispensed in phenytoin sodium equivalent units (PE). 1.5 mg of fosphenytoin sodium is equivalent to 1 mg phenytoin sodium, and is referred to as 1 mg PE. The amount and concentration of fosphenytoin is always expressed in terms of mg of phenytoin sodium equivalents (mg PE).
Do not confuse the concentration of fosphenytoin with the total amount of drug in the vial.
Caution must be used when administering fosphenytoin due to the risk of dosing errors (see WARNINGS ). Medication errors associated with fosphenytoin have resulted in patients receiving the wrong dose of fosphenytoin. Fosphenytoin is marketed in 2 mL vials containing a total of 100 mg PE and 10 mL vials containing a total of 500 mg PE. Both vials contain a concentration of 50 mg PE/mL. Errors have occurred when the concentration of the vial (50 mg PE/mL) was misinterpreted to mean that the total content of the vial was 50 mg PE. These errors have resulted in two- or ten-fold overdoses of fosphenytoin since each of the vials actually contains a total of 100 mg PE or 500 mg PE. In some cases, ten-fold overdoses were associated with fatal outcomes. To help minimize confusion, the prescribed dose of fosphenytoin should always be expressed in milligrams of phenytoin equivalents (mg PE). Additionally, when ordering and storing fosphenytoin, consider displaying the total drug content (i.e., 100 mg PE/ 2 mL or 500 mg PE/ 10 mL) instead of concentration in computer systems, pre-printed orders, and automated dispensing cabinet databases to help ensure that total drug content can be clearly identified. Care should be taken to ensure the appropriate volume of fosphenytoin is withdrawn from the vial when preparing the dose for administration. Attention to these details may prevent some fosphenytoin medication errors from occurring.
Prior to intravenous infusion, dilute fosphenytoin injection in 5% dextrose or 0.9% saline solution for injection to a concentration ranging from 1.5 to 25 mg PE/mL. The maximum concentration of fosphenytoin in any solution should be 25 mg PE/mL. When fosphenytoin is given as an intravenous infusion, fosphenytoin needs to be diluted and should only be administered at a rate not exceeding 150 mg PE/min.
Parenteral drug products should be inspected visually for particulate matter or discoloration prior to administration, whenever solution and container permit.
Status Epilepticus
- The loading dose of fosphenytoin is 15 to 20 mg PE/kg administered at 100 to 150 mg PE/min.
- Because of the risk of hypotension, fosphenytoin should be administered no faster than 150 mg PE/min. Continuous monitoring of the electrocardiogram, blood pressure, and respiratory function is essential and the patient should be observed throughout the period where maximal serum phenytoin concentrations occur, approximately 10 to 20 minutes after the end of fosphenytoin infusions.
- Because the full antiepileptic effect of phenytoin, whether given as fosphenytoin or parenteral phenytoin, is not immediate, other measures, including concomitant administration of an intravenous benzodiazepine, will usually be necessary for the control of status epilepticus.
- The loading dose should be followed by maintenance doses of either fosphenytoin or phenytoin.
If administration of fosphenytoin does not terminate seizures, the use of other anticonvulsants and other appropriate measures should be considered.
Even though loading doses of fosphenytoin have been given by the intramuscular route for other indications when intravenous access is impossible, intramuscular fosphenytoin should ordinarily not be used in the treatment of status epilepticus because therapeutic phenytoin concentrations may not be reached as quickly as with intravenous administration.
Nonemergent Loading and Maintenance Dosing
Because of the risks of cardiac and local toxicity associated with intravenous fosphenytoin, oral phenytoin should be used whenever possible.
The loading dose of fosphenytoin is 10 to 20 mg PE/kg given intravenous or intramuscular. The rate of administration for intravenous fosphenytoin should be no greater than 150 mg PE/min. Continuous monitoring of the electrocardiogram, blood pressure, and respiratory function is essential and the patient should be observed throughout the period where maximal serum phenytoin concentrations occur (approximately 20 minutes after the end of fosphenytoin infusion).
The initial daily maintenance dose of fosphenytoin is 4 to 6 mg PE/kg/day in divided doses.
Intramuscular or Intravenous Substitution for Oral Phenytoin Therapy
When treatment with oral phenytoin is not possible, fosphenytoin can be substituted for oral phenytoin at the same total daily dose.
Phenytoin sodium capsules are approximately 90% bioavailable by the oral route. Phenytoin, supplied as fosphenytoin, is 100% bioavailable by both the intramuscular and intravenous routes. For this reason, plasma phenytoin concentrations may increase modestly when intramuscular or intravenous fosphenytoin is substituted for oral phenytoin sodium therapy.
The rate of administration for intravenous fosphenytoin should be no greater than 150 mg PE/min.
In controlled trials, intramuscular fosphenytoin was administered as a single daily dose utilizing either 1 or 2 injection sites. Some patients may require more frequent dosing.
Dosing in Special Populations
Patients with Renal or Hepatic Disease: Due to an increased fraction of unbound phenytoin in patients with renal or hepatic disease, or in those with hypoalbuminemia, the interpretation of total phenytoin plasma concentrations should be made with caution (see CLINICAL PHARMACOLOGY, Special Populations ). Unbound phenytoin concentrations may be more useful in these patient populations. After intravenous fosphenytoin administration to patients with renal and/or hepatic disease, or in those with hypoalbuminemia, fosphenytoin clearance to phenytoin may be increased without a similar increase in phenytoin clearance. This has the potential to increase the frequency and severity of adverse events (see PRECAUTIONS ).
Elderly Patients: Age does not have a significant impact on the pharmacokinetics of fosphenytoin following fosphenytoin administration. Phenytoin clearance is decreased slightly in elderly patients and lower or less frequent dosing may be required.
Pediatric: The safety and efficacy of fosphenytoin in pediatric patients have not been established.
CONTRAINDICATIONS
Fosphenytoin sodium injection, USP is contraindicated in patients who have demonstrated hypersensitivity to fosphenytoin sodium injection, USP or its ingredients, or to phenytoin or other hydantoins.
Because of the effect of parenteral phenytoin on ventricular automaticity, fosphenytoin injection is contraindicated in patients with sinus bradycardia, sino-atrial block, second and third degree A-V block, and Adams-Stokes syndrome.
Coadministration of fosphenytoin is contraindicated with delavirdine due to potential for loss of virologic response and possible resistance to delavirdine or to the class of non-nucleoside reverse transcriptase inhibitors.
ADVERSE REACTIONS
The more important adverse clinical events caused by the IV use of fosphenytoin or phenytoin are cardiovascular collapse and/or central nervous system depression. Hypotension can occur when either drug is administered rapidly by the IV route. The rate of administration is very important; for fosphenytoin, it should not exceed 150 mg PE/min.
The adverse clinical events most commonly observed with the use of fosphenytoin in clinical trials were nystagmus, dizziness, pruritus, paresthesia, headache, somnolence, and ataxia. With two exceptions, these events are commonly associated with the administration of IV phenytoin. Paresthesia and pruritus, however, were seen much more often following fosphenytoin administration and occurred more often with IV fosphenytoin administration than with IM fosphenytoin administration. These events were dose and rate related; most alert patients (41 of 64; 64%) administered doses of ≥15 mg PE/kg at 150 mg PE/min experienced discomfort of some degree. These sensations, generally described as itching, burning, or tingling, were usually not at the infusion site. The location of the discomfort varied with the groin mentioned most frequently as a site of involvement. The paresthesia and pruritus were transient events that occurred within several minutes of the start of infusion and generally resolved within 10 minutes after completion of fosphenytoin infusion. Some patients experienced symptoms for hours. These events did not increase in severity with repeated administration. Concurrent adverse events or clinical laboratory change suggesting an allergic process were not seen (see PRECAUTIONS, Sensory Disturbances ).
Approximately 2% of the 859 individuals who received fosphenytoin in premarketing clinical trials discontinued treatment because of an adverse event. The adverse events most commonly associated with withdrawal were pruritus (0.5%), hypotension (0.3%), and bradycardia (0.2%).
Dose and Rate Dependency of Adverse Events Following IV Fosphenytoin :
The incidence of adverse events tended to increase as both dose and infusion rate increased. In particular, at doses of ≥15 mg PE/kg and rates ≥150 mg PE/min, transient pruritus, tinnitus, nystagmus, somnolence, and ataxia occurred 2 to 3 times more often than at lower doses or rates.
Incidence in Controlled Clinical Trials
All adverse events were recorded during the trials by the clinical investigators using terminology of their own choosing. Similar types of events were grouped into standardized categories using modified COSTART dictionary terminology. These categories are used in the tables and listings below with the frequencies representing the proportion of individuals exposed to fosphenytoin or comparative therapy.
The prescriber should be aware that these figures cannot be used to predict the frequency of adverse events in the course of usual medical practice where patient characteristics and other factors may differ from those prevailing during clinical studies. Similarly, the cited frequencies cannot be directly compared with figures obtained from other clinical investigations involving different treatments, uses or investigators. An inspection of these frequencies, however, does provide the prescribing physician with one basis to estimate the relative contribution of drug and nondrug factors to the adverse event incidences in the population studied.
Incidence in Controlled Clinical Trials - IV Administration To Patients With Epilepsy or Neurosurgical Patients : Table 2 lists treatment-emergent adverse events that occurred in at least 2% of patients treated with IV fosphenytoin at the maximum dose and rate in a randomized, double-blind, controlled clinical trial where the rates for phenytoin and fosphenytoin administration would have resulted in equivalent systemic exposure to phenytoin.
| TABLE 2. Treatment-Emergent Adverse Event Incidence Following IV Administration at the Maximum Dose and Rate to Patients with Epilepsy or Neurosurgical Patients (Events in at Least 2% of Fosphenytoin-Treated Patients) | ||
| BODY SYSTEM Adverse Event | IV Fosphenytoin N=90 | IV Phenytoin N=22 |
| BODY AS A WHOLE | ||
| Pelvic Pain | 4.4 | 0 |
| Asthenia | 2.2 | 0 |
| Back Pain | 2.2 | 0 |
| Headache | 2.2 | 4.5 |
| CARDIOVASCULAR | ||
| Hypotension | 7.7 | 9.1 |
| Vasodilatation | 5.6 | 4.5 |
| Tachycardia | 2.2 | 0 |
| DIGESTIVE | ||
| Nausea | 8.9 | 13.6 |
| Tongue Disorder | 4.4 | 0 |
| Dry Mouth | 4.4 | 4.5 |
| Vomiting | 2.2 | 9.1 |
| NERVOUS | ||
| Nystagmus | 44.4 | 59.1 |
| Dizziness | 31.1 | 27.3 |
| Somnolence | 20 | 27.3 |
| Ataxia | 11.1 | 18.2 |
| Stupor | 7.7 | 4.5 |
| Incoordination | 4.4 | 4.5 |
| Paresthesia | 4.4 | 0 |
| Extrapyramidal Syndrome | 4.4 | 0 |
| Tremor | 3.3 | 9.1 |
| Agitation | 3.3 | 0 |
| Hypesthesia | 2.2 | 9.1 |
| Dysarthria | 2.2 | 0 |
| Vertigo | 2.2 | 0 |
| Brain Edema | 2.2 | 4.5 |
| SKIN AND APPENDAGES | ||
| Pruritus | 48.9 | 4.5 |
| SPECIAL SENSES | ||
| Tinnitus | 8.9 | 9.1 |
| Diplopia | 3.3 | 0 |
| Taste Perversion | 3.3 | 0 |
| Amblyopia | 2.2 | 9.1 |
| Deafness | 2.2 | 0 |
Incidence in Controlled Trials - IM Administration To Patients With Epilepsy: Table 3 lists treatment-emergent adverse events that occurred in at least 2% of fosphenytoin-treated patients in a double-blind, randomized, controlled clinical trial of adult epilepsy patients receiving either IM fosphenytoin substituted for oral phenytoin sodium or continuing oral phenytoin sodium. Both treatments were administered for 5 days.
| TABLE 3. Treatment-Emergent Adverse Event Incidence Following Substitution of IM Fosphenytoin for Oral Phenytoin Sodium in Patients With Epilepsy (Events in at Least 2% of Fosphenytoin-Treated Patients) | ||
| BODY SYSTEM Adverse Event | IM Fosphenytoin N=179 | Oral Phenytoin Sodium N=61 |
| BODY AS A WHOLE | ||
| Headache | 8.9 | 4.9 |
| Asthenia | 3.9 | 3.3 |
| Accidental Injury | 3.4 | 6.6 |
| DIGESTIVE | ||
| Nausea | 4.5 | 0 |
| Vomiting | 2.8 | 0 |
| HEMATOLOGIC AND LYMPHATIC | ||
| Ecchymosis | 7.3 | 4.9 |
| NERVOUS | ||
| Nystagmus | 15.1 | 8.2 |
| Tremor | 9.5 | 13.1 |
| Ataxia | 8.4 | 8.2 |
| Incoordination | 7.8 | 4.9 |
| Somnolence | 6.7 | 9.8 |
| Dizziness | 5 | 3.3 |
| Paresthesia | 3.9 | 3.3 |
| Reflexes Decreased | 2.8 | 4.9 |
| SKIN AND APPENDAGES | ||
| Pruritus | 2.8 | 0 |
Adverse Events During All Clinical Trials
Fosphenytoin has been administered to 859 individuals during all clinical trials. All adverse events seen at least twice are listed in the following, except those already included in previous tables and listings. Events are further classified within body system categories and enumerated in order of decreasing frequency using the following definitions: frequent adverse events are defined as those occurring in greater than 1/100 individuals; infrequent adverse events are those occurring in 1/100 to 1/1000 individuals.
Body as a Whole: Frequent : fever, injection-site reaction, infection, chills, face edema, injection-site pain; Infrequent : sepsis, injection-site inflammation, injection-site edema, injection-site hemorrhage, flu syndrome, malaise, generalized edema, shock, photosensitivity reaction, cachexia, cryptococcosis.
Cardiovascular : Frequent : hypertension; Infrequent : cardiac arrest, migraine, syncope, cerebral hemorrhage, palpitation, sinus bradycardia, atrial flutter, bundle branch block, cardiomegaly, cerebral infarct, postural hypotension, pulmonary embolus, QT interval prolongation, thrombophlebitis, ventricular extrasystoles, congestive heart failure.
Digestive : Frequent : constipation; Infrequent : dyspepsia, diarrhea, anorexia, gastrointestinal hemorrhage, increased salivation, liver function tests abnormal, tenesmus, tongue edema, dysphagia, flatulence, gastritis, ileus.
Endocrine : Infrequent : diabetes insipidus.
Hematologic and Lymphatic: Infrequent : thrombocytopenia, anemia, leukocytosis, cyanosis,
hypochromic anemia, leukopenia, lymphadenopathy, petechia.
Metabolic and Nutritional: Frequent : hypokalemia; Infrequent : hyperglycemia, hypophosphatemia, alkalosis, acidosis, dehydration, hyperkalemia, ketosis.
Musculoskeletal: Frequent : myasthenia; Infrequent : myopathy, leg cramps, arthralgia, myalgia.
Nervous: Frequent : reflexes increased, speech disorder, dysarthria, intracranial hypertension, thinking abnormal, nervousness, hypesthesia; Infrequent : confusion, twitching, Babinski sign positive, circumoral paresthesia, hemiplegia, hypotonia, convulsion, extrapyramidal syndrome, insomnia, meningitis, depersonalization, CNS depression, depression, hypokinesia, hyperkinesia, brain edema, paralysis, psychosis, aphasia, emotional lability, coma, hyperesthesia, myoclonus, personality disorder, acute brain syndrome, encephalitis, subdural hematoma, encephalopathy, hostility, akathisia, amnesia, neurosis.
Respiratory : Frequent : pneumonia; Infrequent : pharyngitis, sinusitis, hyperventilation, rhinitis, apnea, aspiration pneumonia, asthma, dyspnea, atelectasis, cough increased, sputum increased, epistaxis, hypoxia, pneumothorax, hemoptysis, bronchitis.
Skin and Appendages : Frequent : rash; Infrequent : maculopapular rash, urticaria, sweating, skin discoloration, contact dermatitis, pustular rash, skin nodule.
Special Senses: Frequent : taste perversion; Infrequent : deafness, visual field defect, eye pain, conjunctivitis, photophobia, hyperacusis, mydriasis, parosmia, ear pain, taste loss.
Urogenital: Infrequent : urinary retention, oliguria, dysuria, vaginitis, albuminuria, genital edema, kidney failure, polyuria, urethral pain, urinary incontinence, vaginal moniliasis.
Post-Marketing Experience
The following adverse reactions have been identified during postapproval use of fosphenytoin. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
There have been post-marketing reports of anaphylactoid reaction and anaphylaxis.
Other Phenytoin-Associated Adverse Events:
Dyskinesia.
Drug Interactions
No drugs are known to interfere with the conversion of fosphenytoin to phenytoin. Conversion could be affected by alterations in the level of phosphatase activity, but given the abundance and wide distribution of phosphatases in the body it is unlikely that drugs would affect this activity enough to affect conversion of fosphenytoin to phenytoin. Drugs highly bound to albumin could increase the unbound fraction of fosphenytoin. Although, it is unknown whether this could result in clinically significant effects, caution is advised when administering fosphenytoin with other drugs that significantly bind to serum albumin.
The pharmacokinetics and protein binding of fosphenytoin, phenytoin, and diazepam were not altered when diazepam and fosphenytoin were concurrently administered in single submaximal doses.
The most significant drug interactions following administration of fosphenytoin are expected to occur with drugs that interact with phenytoin. Phenytoin is extensively bound to serum plasma proteins and is prone to competitive displacement. Phenytoin is metabolized by hepatic cytochrome P450 enzymes CYP2C9 and CYP2C19 and is particularly susceptible to inhibitory drug interactions because it is subject to saturable metabolism. Inhibition of metabolism may produce significant increases in circulating phenytoin concentrations and enhance the risk of drug toxicity. Phenytoin is a potent inducer of hepatic drug-metabolizing enzymes.
The most commonly occurring drug interactions are listed below:
Note: The list is not intended to be inclusive or comprehensive. Individual drug package inserts should be consulted.
Drugs that affect phenytoin concentrations:
- Drugs that may increase plasma phenytoin concentrations include: acute alcohol intake, amiodarone, anti-epileptic agents (ethosuximide, felbamate, oxcarbazepine, methsuximide, topiramate), azoles (fluconazole, ketoconazole, itraconazole, miconazole, voriconazole), capecitabine, chloramphenicol, chlordiazepoxide, disulfiram, estrogens, fluorouracil, fluoxetine, fluvastatin, fluvoxamine, H 2 -antagonists (e.g. cimetidine), halothane, isoniazid, methylphenidate, omeprazole, phenothiazines, salicylates, sertraline, succinimides, sulfonamides (e.g., sulfamethizole, sulfaphenazole, sulfadiazine, sulfamethoxazole-trimethoprim), tacrolimus, ticlopidine, tolbutamide, trazodone and warfarin.
- Drugs that may decrease plasma phenytoin concentrations include: anticancer drugs usually in combination (e.g., bleomycin, carboplatin, cisplatin, doxorubicin, methotrexate), carbamazepine, chronic alcohol abuse, diazepam, diazoxide, folic acid, fosamprenavir, nelfinavir, reserpine, rifampin, ritonavir, St. John’s Wort, theophylline and vigabatrin.
- Drugs that may either increase or decrease plasma phenytoin concentrations include: phenobarbital, valproic acid and sodium valproate. Similarly, the effects of phenytoin on phenobarbital, valproic acid and sodium plasma valproate concentrations are unpredictable.
- The addition or withdrawal of these agents in patients on phenytoin therapy may require an adjustment of the phenytoin dose to achieve optimal clinical outcome.
Drugs affected by phenytoin:
- Drugs that should not be coadministered with phenytoin: Delavirdine (see CONTRAINDICATIONS ).
- Drugs whose efficacy is impaired by phenytoin include: azoles (fluconazole, ketoconazole, itraconazole, voriconazole, posaconazole), corticosteroids, doxycycline, estrogens, furosemide, irinotecan, oral contraceptives, paclitaxel, paroxetine, quinidine, rifampin, sertraline, teniposide, theophylline and vitamin D.
- Increased and decreased PT/INR responses have been reported when phenytoin is coadministered with warfarin.
- Phenytoin decreases plasma concentrations of active metabolites of albendazole, certain HIV antivirals (efavirenz, lopinavir/ritonavir, indinavir, nelfinavir, ritonavir, saquinavir), anti-epileptic agents (carbamazepine, felbamate, lamotrigine, topiramate, oxcarbazepine, quetiapine), atorvastatin, chlorpropamide, clozapine, cyclosporine, digoxin, fluvastatin, folic acid, methadone, mexiletine, nifedipine, nimodipine, nisoldipine, praziquantel, simvastatin and verapamil.
- Phenytoin when given with fosamprenavir alone may decrease the concentration of amprenavir, the active metabolite. Phenytoin when given with the combination of fosamprenavir and ritonavir may increase the concentration of amprenavir.
- Resistance to the neuromuscular blocking action of the nondepolarizing neuromuscular blocking agents pancuronium, vecuronium, rocuronium and cisatracurium has occurred in patients chronically administered phenytoin. Whether or not phenytoin has the same effect on other non-depolarizing agents is unknown. Patients should be monitored closely for more rapid recovery from neuromuscular blockade than expected, and infusion rate requirements may be higher.
- The addition or withdrawal of phenytoin during concomitant therapy with these agents may require adjustment of the dose of these agents to achieve optimal clinical outcome.
Monitoring of plasma phenytoin concentrations may be helpful when possible drug interactions are suspected (see Laboratory Tests ).
DESCRIPTION
Fosphenytoin sodium injection, USP is a prodrug intended for parenteral administration; its active metabolite is phenytoin. 1.5 mg fosphenytoin sodium, USP (hereafter referred to as fosphenytoin) equivalent to 1 mg phenytoin sodium and is referred to as 1 mg phenytoin equivalents (PE). The amount and concentration of fosphenytoin is always expressed in terms of mg PE.
Fosphenytoin injection is marketed in 2 mL vials containing a total of 100 mg PE and 10 mL vials containing a total of 500 mg PE. The concentration of each vial is 50 mg PE/mL. Fosphenytoin is supplied in vials as a ready-mixed solution in Water for Injection, USP, and Tromethamine, USP (TRIS), buffer adjusted to pH 8.6 to 9.0 with either Hydrochloric Acid, NF, or Sodium Hydroxide, NF. Fosphenytoin injection is a clear, colorless to pale yellow, sterile solution.
The chemical name of fosphenytoin is 5,5-diphenyl-3-[(phosphonooxy)methyl]-2,4-imidazolidinedione disodium salt.
The molecular structure of fosphenytoin is:
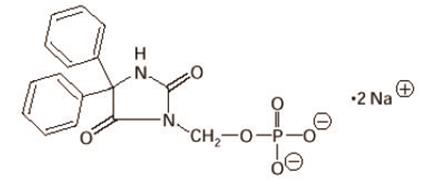
The molecular weight of fosphenytoin is 406.24.
IMPORTANT NOTE: Throughout all fosphenytoin injection product labeling, the amount and concentration of fosphenytoin are always expressed in terms of phenytoin sodium equivalents (PE). Fosphenytoin’s weight is expressed as phenytoin sodium equivalents to avoid the need to perform molecular weight-based adjustments when substituting fosphenytoin for phenytoin or vice versa.
Care should be taken to ensure that fosphenytoin is always prescribed and dispensed in phenytoin sodium equivalents (PE) (see DOSAGE AND ADMINISTRATION ).
CLINICAL PHARMACOLOGY
Introduction
Following parenteral administration of fosphenytoin injection, fosphenytoin is converted to the anticonvulsant phenytoin. For every mmol of fosphenytoin administered, one mmol of phenytoin is produced. The pharmacological and toxicological effects of fosphenytoin include those of phenytoin. However, the hydrolysis of fosphenytoin to phenytoin yields two metabolites, phosphate and formaldehyde. Formaldehyde is subsequently converted to formate, which is in turn metabolized via a folate dependent mechanism. Although phosphate and formaldehyde (formate) have potentially important biological effects, these effects typically occur at concentrations considerably in excess of those obtained when fosphenytoin is administered under conditions of use recommended in this labeling.
Mechanism of Action
Fosphenytoin is a prodrug of phenytoin and accordingly, its anticonvulsant effects are attributable to phenytoin.
After IV administration to mice, fosphenytoin blocked the tonic phase of maximal electroshock seizures at doses equivalent to those effective for phenytoin. In addition to its ability to suppress maximal electroshock seizures in mice and rats, phenytoin exhibits anticonvulsant activity against kindled seizures in rats, audiogenic seizures in mice, and seizures produced by electrical stimulation of the brainstem in rats. The cellular mechanisms of phenytoin thought to be responsible for its anticonvulsant actions include modulation of voltage-dependent sodium channels of neurons, inhibition of calcium flux across neuronal membranes, modulation of voltage-dependent calcium channels of neurons, and enhancement of the sodium-potassium ATPase activity of neurons and glial cells. The modulation of sodium channels may be a primary anticonvulsant mechanism because this property is shared with several other anticonvulsants in addition to phenytoin.
Pharmacokinetics and Drug Metabolism
Fosphenytoin
Absorption/Bioavailability:
Intravenous: When fosphenytoin is administered by IV infusion, maximum plasma fosphenytoin concentrations are achieved at the end of the infusion. Fosphenytoin has a half-life of approximately 15 minutes.
Intramuscular : Fosphenytoin is completely bioavailable following IM administration of fosphenytoin. Peak concentrations occur at approximately 30 minutes postdose. Plasma fosphenytoin concentrations following IM administration are lower but more sustained than those following IV administration due to the time required for absorption of fosphenytoin from the injection site.
Distribution: Fosphenytoin is extensively bound (95% to 99%) to human plasma proteins, primarily albumin. Binding to plasma proteins is saturable with the result that the percent bound decreases as total fosphenytoin concentrations increase. Fosphenytoin displaces phenytoin from protein binding sites. The volume of distribution of fosphenytoin increases with fosphenytoin dose and rate, and ranges from 4.3 to 10.8 liters.
Metabolism and Elimination: The conversion half-life of fosphenytoin to phenytoin is approximately 15 minutes. The mechanism of fosphenytoin conversion has not been determined, but phosphatases probably play a major role. Fosphenytoin is not excreted in urine. Each mmol of fosphenytoin is metabolized to 1 mmol of phenytoin, phosphate, and formate (see CLINICAL PHARMACOLOGY, Introduction and PRECAUTIONS, Phosphate Load for Renally Impaired Patients ).
Phenytoin (after fosphenytoin administration)
In general, IM administration of fosphenytoin generates systemic phenytoin concentrations that are similar enough to oral phenytoin sodium to allow essentially interchangeable use.
The pharmacokinetics of fosphenytoin following IV administration of fosphenytoin, however, are complex, and when used in an emergency setting (e.g., status epilepticus), differences in rate of availability of phenytoin could be critical. Studies have therefore empirically determined an infusion rate for fosphenytoin that gives a rate and extent of phenytoin systemic availability similar to that of a 50 mg/min phenytoin sodium infusion.
A dose of 15 to 20 mg PE/kg of fosphenytoin infused at 100 to 150 mg PE/min yields plasma free phenytoin concentrations over time that approximate those achieved when an equivalent dose of phenytoin sodium (e.g., parenteral phenytoin sodium) is administered at 50 mg/min (see DOSAGE AND ADMINISTRATION and WARNINGS ).
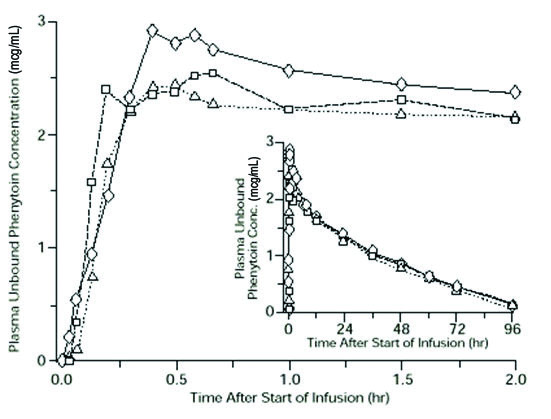
FIGURE 1. Mean plasma unbound phenytoin concentrations following IV administration of 1200 mg PE fosphenytoin infused at 100 mg PE/min (triangles) or 150 mg PE/min (squares) and 1200 mg phenytoin sodium infused at 50 mg/min (diamonds) to healthy subjects (N = 12). Inset shows time course for the entire 96-hour sampling period.
Following administration of single IV fosphenytoin doses of 400 to 1200 mg PE, mean maximum total phenytoin concentrations increase in proportion to dose, but do not change appreciably with changes in infusion rate. In contrast, mean maximum unbound phenytoin concentrations increase with both dose and rate.
Absorption/Bioavailability: Fosphenytoin is completely converted to phenytoin following IV administration, with a half-life of approximately 15 minutes. Fosphenytoin is also completely converted to phenytoin following IM administration and plasma total phenytoin concentrations peak in approximately 3 hours.
Distribution: Phenytoin is highly bound to plasma proteins, primarily albumin, although to a lesser extent than fosphenytoin. In the absence of fosphenytoin, approximately 12% of total plasma phenytoin is unbound over the clinically relevant concentration range. However, fosphenytoin displaces phenytoin from plasma protein binding sites. This increases the fraction of phenytoin unbound (up to 30% unbound) during the period required for conversion of fosphenytoin to phenytoin (approximately 0.5 to 1 hour post infusion).
Metabolism and Elimination: Phenytoin derived from administration of fosphenytoin is extensively metabolized in the liver and excreted in urine primarily as 5-(p-hydroxyphenyl)-5-phenylhydantoin and its glucuronide; little unchanged phenytoin (1% to 5% of the fosphenytoin dose) is recovered in urine. Phenytoin is metabolized by the cytochrome P450 enzymes CYP2C9 and CYP2C19. Phenytoin hepatic metabolism is saturable, and following administration of single IV fosphenytoin doses of 400 to 1200 mg PE, total and unbound phenytoin AUC values increase disproportionately with dose. Mean total phenytoin half-life values (12 to 28.9 hr) following fosphenytoin injection administration at these doses are similar to those after equal doses of parenteral phenytoin sodium and tend to be greater at higher plasma phenytoin concentrations.
Special Populations
Patients with Renal or Hepatic Disease : Due to an increased fraction of unbound phenytoin in patients with renal or hepatic disease, or in those with hypoalbuminemia, the interpretation of total phenytoin plasma concentrations should be made with caution (see DOSAGE AND ADMINISTRATION ). Unbound phenytoin concentrations may be more useful in these patient populations. After IV administration of fosphenytoin to patients with renal and/or hepatic disease, or in those with hypoalbuminemia, fosphenytoin clearance to phenytoin may be increased without a similar increase in phenytoin clearance. This has the potential to increase the frequency and severity of adverse events (see PRECAUTIONS ).
Age : The effect of age was evaluated in patients 5 to 98 years of age. Patient age had no significant impact on fosphenytoin pharmacokinetics. Phenytoin clearance tends to decrease with increasing age (20% less in patients over 70 years of age relative to that in patients 20 to 30 years of age). Phenytoin dosing requirements are highly variable and must be individualized (see DOSAGE AND ADMINISTRATION ).
Gender and Race : Gender and race have no significant impact on fosphenytoin or phenytoin pharmacokinetics.
Pediatrics: The safety and efficacy of fosphenytoin in pediatric patients have not been established.
Clinical Studies
Infusion tolerance was evaluated in clinical studies. One double-blind study assessed infusion-site tolerance of equivalent loading doses (15 to 20 mg PE/kg) of fosphenytoin infused at 150 mg PE/min or phenytoin infused at 50 mg/min. The study demonstrated better local tolerance (pain and burning at the infusion site), fewer disruptions of the infusion, and a shorter infusion period for fosphenytoin-treated patients (Table 1).
| TABLE 1. Infusion Tolerance of Equivalent Loading Doses of IV Fosphenytoin and IV Phenytoin | ||
| IV Fosphenytoin N=90 | IV Phenytoin N=22 | |
| Local Intolerance | 9% a | 90% |
| Infusion Disrupted | 21% | 67% |
| Average Infusion Time | 13 min | 44 min |
| a Percent of patients | ||
Fosphenytoin-treated patients, however, experienced more systemic sensory disturbances (see PRECAUTIONS, Sensory Disturbances ).
Infusion disruptions in fosphenytoin-treated patients were primarily due to systemic burning, pruritus, and/or paresthesia while those in phenytoin-treated patients were primarily due to pain and burning at the infusion site (see Table 1 ).
In a double-blind study investigating temporary substitution of fosphenytoin for oral phenytoin, IM fosphenytoin was as well-tolerated as IM placebo. IM fosphenytoin resulted in a slight increase in transient, mild to moderate local itching (23% of patients vs 11% of IM placebo-treated patients at any time during the study). This study also demonstrated that equimolar doses of IM fosphenytoin may be substituted for oral phenytoin sodium with no dosage adjustments needed when initiating IM or returning to oral therapy. In contrast, switching between IM and oral phenytoin requires dosage adjustments because of slow and erratic phenytoin absorption from muscle.
HOW SUPPLIED
Fosphenytoin Sodium Injection, USP is supplied as follows:
10 mL per vial [NDC 65162-999-01] — Each 10 mL vial contains Fosphenytoin Sodium, USP 750 mg equivalent to 500 mg of phenytoin sodium, USP: (Packages of 10 - NDC 65162-999-10).
2 mL per vial [NDC 65162-998-01] — Each 2 mL vial contains Fosphenytoin Sodium, USP 150 mg equivalent to 100 mg of phenytoin sodium, USP: (Packages of 25 - NDC 65162-998-25).
Both sizes of vials contain Tromethamine, USP (TRIS), Hydrochloric Acid, NF, or Sodium Hydroxide, NF, and Water for Injection, USP.
Fosphenytoin sodium should always be prescribed in phenytoin sodium equivalents (PE) (see DOSAGE AND ADMINISTRATION ).
1.5 mg of fosphenytoin sodium is equivalent to 1 mg phenytoin sodium, and is referred to as 1 mg PE. The amount and concentration of fosphenytoin is always expressed in terms of mg of phenytoin sodium equivalents (PE). Fosphenytoin’s weight is expressed as phenytoin sodium equivalents to avoid the need to perform molecular weight-based adjustments when substituting fosphenytoin for phenytoin or vice versa.
Store under refrigeration at 2°C to 8°C (36°F to 46°F). The product should not be stored at room temperature for more than 48 hours. Vials that develop particulate matter should not be used.
Brands listed are the trademarks of their respective owners.
Made in India
Distributed by: Amneal Pharmaceuticals Bridgewater, NJ 08807
Rev. 10-2015-03