Get your patient on Imiquimod - Imiquimod cream (Imiquimod)
Imiquimod - Imiquimod cream prescribing information
Warnings and Precautions, Local Hypopigmentation Reactions (5.2 ) | 09/2024 |
INDICATIONS AND USAGE
Imiquimod Cream is indicated for the topical treatment of:
• Clinically typical, nonhyperkeratotic, nonhypertrophic actinic keratoses (AK) on the face or scalp in immunocompetent adults. (1.1 )
• Biopsy-confirmed, primary superficial basal cell carcinoma (sBCC) in immunocompetent adults with a maximum tumor diameter of 2.0 cm on trunk (excluding anogenital skin), neck, or extremities (excluding hands and feet), only when surgical methods are medically less appropriate and patient follow-up can be reasonably assured. (1.2 )
• External genital and perianal warts (EGW) in immunocompetent patients 12 years of age and older. (1.3 )
Actinic Keratosis
Imiquimod Cream is indicated for the topical treatment of clinically typical, nonhyperkeratotic, nonhypertrophic actinic keratoses (AK) on the face or scalp in immunocompetent adults.
Superficial Basal Cell Carcinoma
Imiquimod Cream is indicated for the topical treatment of biopsy-confirmed, primary superficial basal cell carcinoma (sBCC) in immunocompetent adults, with a maximum tumor diameter of 2.0 cm, located on the trunk (excluding anogenital skin), neck, or extremities (excluding hands and feet), only when surgical methods are medically less appropriate and patient follow-up can be reasonably assured.
Establish the histological diagnosis of superficial basal cell carcinoma prior to treatment. The safety and effectiveness of Imiquimod Cream have not been established for other types of basal cell carcinomas (BCC), including nodular and morpheaform (fibrosing or sclerosing) types.
External Genital Warts
Imiquimod Cream is indicated for the topical treatment of external genital and perianal warts (EGW) in immunocompetent patients 12 years of age and older.
DOSAGE AND ADMINISTRATION
- For topical use only; not for oral, ophthalmic, intra-anal or intravaginal use. (2.1 )
- AK: Apply once daily before bedtime 2 times per week for a full 16 weeks to a contiguous area of approximately 25 cm 2 on the face or scalp. Apply no more than 1 packet at each application. (2.2 )
- sBCC: Apply once daily before bedtime 5 times per week for a full 6 weeks to a target tumor with 2 cm maximum diameter on the trunk (excluding anogenital skin), neck, or extremities (excluding hands and feet). Amount of Imiquimod Cream used based on target tumor diameter. (2.3 )
- EGW: Apply thin layer once daily before bedtime 3 times per week until total clearance or for a maximum of 16 weeks. (2.4 )
Important Dosage and Administration Instructions
Imiquimod Cream is for topical use only. Imiquimod Cream is not for oral, ophthalmic, or intravaginal use. Instruct patients on proper application technique.
Wash hands before and after applying Imiquimod Cream. Wash the treatment area with mild soap and water and allow the area to dry thoroughly (at least 10 minutes) before applying Imiquimod Cream.
If an Imiquimod Cream dose is missed, apply the next dose at the regularly scheduled time.
Avoid contact with the eyes, lips, nostrils, or inside the anus and vagina.
For patients with AK and sBCC, prescribe no more than 3 boxes (36 packets) of Imiquimod Cream for the entire treatment period. For EGW, one packet of Imiquimod Cream contains sufficient cream to cover a wart area of up to 20 cm 2 .
Discard partially used packets and do not reuse.
Dosage and Administration for Actinic Keratosis
Apply Imiquimod Cream topically once daily before bedtime 2 times per week for a full 16 weeks to a defined treatment area of AK on the face or scalp (but not both concurrently). A treatment area is defined as one contiguous area of approximately 25 cm 2 (e.g., 5 cm × 5 cm) on the face (e.g., forehead or one cheek) or on the scalp. Apply Imiquimod Cream to the entire treatment area and rub in until the cream is no longer visible. Apply no more than 1 packet of Imiquimod Cream to the contiguous treatment area at each application. Leave Imiquimod Cream on the skin for approximately 8 hours and then remove with mild soap and water.
For local skin reactions a dosage interruption of several days may be taken if required by the patient's discomfort or severity of the local skin reaction [see Warnings and Precautions (5.1 )] . Do not extend treatment beyond 16 weeks due to missed doses or rest periods. Assess response to treatment after resolution of local skin reactions.
Dosage and Administration for Superficial Basal Cell Carcinoma
Apply Imiquimod Cream topically once daily before bedtime 5 times per week for a full 6 weeks to a biopsy-confirmed sBCC. The target tumor should have a maximum diameter of 2 cm and be located on the trunk (excluding anogenital skin), neck, or extremities (excluding hands and feet). The amount of cream needed to cover the target tumor, including 1 cm of skin surrounding the tumor, is presented in Table 1. Rub Imiquimod Cream into the treatment area until the cream is no longer visible. Leave Imiquimod Cream on the skin for approximately 8 hours and then remove with mild soap and water.
Table 1: Amount of Imiquimod Cream to Use for sBCC
Target Tumor Diameter | Size of Cream Droplet to be Used (Diameter) | Approximate Amount of Imiquimod Cream to be Used |
0.5 to <1.0 cm | 4 mm | 10 mg |
≥1.0 to <1.5 cm | 5 mm | 25 mg |
≥1.5 to 2.0 cm | 7 mm | 40 mg |
For local skin reactions a dosage interruption of several days may be taken if required by the patient's discomfort or severity of the local skin reaction [see Warnings and Precautions (5.1 )] .
Assess for early clinical clearance after resolution of local skin reactions (e.g., 12 weeks post-treatment). Local skin reactions or other findings (e.g., infection) may require that a patient be seen sooner than the post-treatment assessment for clinical clearance. If there is clinical evidence of persistent tumor at the post-treatment assessment for clinical clearance, consider a biopsy or other alternative intervention. Instruct patients to contact their healthcare provider if any suspicious lesion arises in the treatment area at any time after a determination of clinical clearance [see Clinical Studies (14.2 )] .
Dosage and Administration for External Genital Warts
Apply a thin layer of Imiquimod Cream topically once daily before bedtime 3 times per week to EGW until there is total clearance of the genital/perianal warts or for a maximum of 16 weeks. Rub in until the cream is no longer visible. Do not occlude the application site. Leave Imiquimod Cream on the skin for 6 to 10 hours and then remove with mild soap and water.
For local skin reactions, a dosage interruption of several days may be taken if required by the patient's discomfort or severity of the local skin reaction [see Warnings and Precautions (5.1 )] . Treatment may resume once the reaction subsides. Nonocclusive dressings such as cotton gauze or cotton underwear may be used to manage skin reactions.
Inform uncircumcised patients treating warts under the foreskin to retract the foreskin and clean the area daily.
Imiquimod Cream may weaken condoms and vaginal diaphragms; therefore, concurrent use is not recommended.
DOSAGE FORMS AND STRENGTHS
Cream, 5%: a white to off-white cream in unit-dose packets, with each packet containing 250 mg of cream, equivalent to 12.5 mg of imiquimod.
USE IN SPECIFIC POPULATIONS
Pregnancy
Risk Summary
Available data from case reports and case series of use with imiquimod during pregnancy have not identified a drug-associated risk of major birth defects, miscarriage or adverse maternal or fetal outcomes. There are no controlled or large-scale epidemiologic studies and no exposure registries with imiquimod use in pregnant women.
In animal reproduction studies, there were no adverse developmental effects observed after oral administration of imiquimod in pregnant rats and intravenous administration of imiquimod in pregnant rabbits during organogenesis at doses up to 98 times and 407 times, respectively, the maximum recommended human dose (MRHD) (see Data) .
The background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Data
Animal Data
The MRHD was set at 2 packets per treatment of Imiquimod Cream (25 mg imiquimod) for the animal multiples of human exposure presented in this label.
Systemic embryofetal development studies were conducted in rats and rabbits. Oral doses of 1, 5, and 20 mg/kg/day imiquimod were administered during the period of organogenesis to pregnant female rats. In the presence of maternal toxicity, fetal effects noted at 20 mg/kg/day (577 times the MRHD based on AUC comparison) included increased resorptions, decreased fetal body weights, delays in skeletal ossification, bent limb bones, and two fetuses in one litter (2 of 1567 fetuses) demonstrated exencephaly, protruding tongues and low-set ears. No treatment-related effects on embryofetal toxicity or malformation were noted at 5 mg/kg/day (98 times the MRHD based on AUC comparison).
Intravenous doses of 0.5, 1, and 2 mg/kg/day imiquimod were administered during the period of organogenesis to pregnant female rabbits. No treatment-related effects on embryofetal toxicity or malformation were noted at 2 mg/kg/day (1.5 times the MRHD based on BSA comparison), the highest dose evaluated in this study, or 1 mg/kg/day (407 times the MRHD based on AUC comparison).
A combined fertility and peri- and postnatal development study was conducted in rats. Oral doses of 1, 1.5, 3, and 6 mg/kg/day imiquimod were administered to male rats from 70 days prior to mating through the mating period and to female rats from 14 days prior to mating through parturition and lactation. No effects on growth, fertility, reproduction, or postnatal development were noted at doses up to 6 mg/kg/day (87 times the MRHD based on AUC comparison), the highest dose evaluated in this study. In the absence of maternal toxicity, bent limb bones were noted in the F1 fetuses at a dose of 6 mg/kg/day (87 times the MRHD based on AUC comparison). This fetal effect was also noted in the oral rat embryofetal development study conducted with imiquimod. No treatment-related malformations were noted at 3 mg/kg/day (41 times the MRHD based on AUC comparison).
Lactation
Risk Summary
There is no information available on the presence of imiquimod in human milk, the effects of the drug on the breastfed infant, or the effects of the drug on milk production after topical application of Imiquimod Cream to women who are breastfeeding. Systemic concentration following topical administration of imiquimod cream is low; therefore, transfer of Imiquimod Cream into breastmilk is likely to be low [see Clinical Pharmacology (12.3)] . The development and health benefits of breastfeeding should be considered along with the mother’s clinical need for Imiquimod Cream and any potential adverse effects on the breastfed infant from Imiquimod Cream or from the underlying maternal condition.
Clinical Considerations
Avoid application of Imiquimod Cream to areas with increased risk for potential ingestion by or ocular exposure to the breastfeeding child.
Pediatric Use
Actinic Keratosis and Superficial Basal Cell Carcinoma
The safety and effectiveness of Imiquimod Cream for the treatment of AK or sBCC in pediatric patients have not been established.
External Genital Warts
The safety and effectiveness of Imiquimod Cream for the treatment of EGW in pediatric patients 12 years of age and older have been established. Use of Imiquimod Cream for this indication is supported by evidence from adequate and well controlled trials in adults [see Clinical Studies (14.3)] . The safety and effectiveness of Imiquimod Cream for the treatment of EGW in pediatric patients less than 12 years of age have not been established.
Molluscum Contagiosum
The safety and effectiveness of Imiquimod Cream for the treatment of molluscum contagiosum (MC) in pediatric patients have not been established. Safety and effectiveness of Imiquimod Cream was not demonstrated in two randomized, vehicle-controlled, double-blind trials involving 702 pediatric subjects with MC (470 exposed to Imiquimod Cream; median age 5 years, range 2–12 years).
Adverse reactions reported in pediatric subject with MC (and not previously reported) included otitis media (5% Imiquimod Cream vs. 3% vehicle) and conjunctivitis (3% Imiquimod Cream vs. 2% vehicle).
In a pharmacokinetics trial in subjects aged 2 to 12 years with extensive MC involving at least 10% of total body surface area; among the 20 subjects with evaluable laboratory assessments, the median white blood cell (WBC) count decreased by 1.4 x 10 9 /L and the median absolute neutrophil count decreased by 1.42 x 10 9 /L.
Geriatric Use
Of the 215 subjects treated with imiquimod cream in the AK clinical trials, 127 subjects (59%) were 65 years of age or older, while 60 subjects (28%) were 75 years of age or older. Of the 185 subjects treated with imiquimod cream in the sBCC clinical trials, 65 subjects (35%) were 65 years of age or older, while 25 subjects (14%) were 75 years of age or older. No overall differences in safety or effectiveness of Imiquimod Cream have been observed between subjects 65 years of age and older and younger adult subjects.
CONTRAINDICATIONS
None.
WARNINGS AND PRECAUTIONS
- Local Skin Reactions: Intense local inflammatory reactions can occur (e.g., skin weeping, erosion) Dosage interruption may be required. Severe vulvar swelling may occur and lead to urinary retention; interrupt dosing or discontinue for severe vulvar swelling. (5.1 )
- Local Hypopigmentation Reactions: Localized complete depigmentation has occurred and persisted. Discontinue if hypopigmentation develops. (5.2 )
- Systemic Reactions: Flu-like systemic signs and symptoms have occurred. Consider dosage interruption for systemic reactions. (5.3 )
- Ultraviolet Light Exposure Risks: Avoid or minimize exposure to sunlight and sunlamps. Wear sunscreen and protective clothing. (5.4 )
Local Skin Reactions
Local skin reactions including skin weeping or erosion have been reported with Imiquimod Cream and can occur after a few applications [see Adverse Reactions (6.1 )] . Concomitant use of Imiquimod Cream and any other imiquimod products, in the same treatment area, may increase the risk for and severity of local skin reactions.
Imiquimod Cream has the potential to exacerbate inflammatory conditions of the skin, including chronic graft versus host disease.
Severe local inflammatory reactions of the female external genitalia can lead to severe vulvar swelling and urinary retention.
Avoid sexual (genital, anal, oral) contact while Imiquimod Cream is on the skin.
To reduce the risk of local skin reactions and manage local skin reactions that occur with Imiquimod Cream treatment:
- Avoid concomitant use of Imiquimod Cream with any other imiquimod product in the same treatment area.
- Avoid application of Imiquimod Cream to skin that is not intact (i.e., any area with an abrasion, cut, burn, rash, infection, or other condition that has altered skin integrity).
- An interruption of dosing may be required for local skin reactions [see Dosage and Administration (2.2 , 2.3 , 2.4 )] . Interrupt dosing or discontinue Imiquimod Cream for severe vulvar swelling [see Dosage and Administration (2.4 )] .
- If severe local skin reactions occur, instruct patients to remove Imiquimod Cream by washing the treatment area with mild soap and water.
Local Hypopigmentation Reactions
Cases of hypopigmentation, including complete depigmentation, were reported during postmarketing use of Imiquimod Cream. In some cases, hypopigmentation and complete depigmentation did not improve or resolve with treatment and persisted for up to 60 months at the time of reporting. Discontinue Imiquimod Cream if hypopigmentation develops.
Systemic Reactions
Flu-like signs and symptoms have been reported with use of Imiquimod Cream and may accompany, or even precede, local inflammatory reactions [see Adverse Reactions (6.1 )] . Signs and symptoms may include malaise, fever, nausea, myalgias, and rigors. Concomitant use of Imiquimod Cream and any other imiquimod products may increase the risk for and severity of systemic reactions. Consider an interruption of dosing if systemic reactions occur.
Ultraviolet Light Exposure Risks
Imiquimod Cream may cause heightened sunburn susceptibility. Avoid or minimize exposure to sunlight (including sunlamps) during use of Imiquimod Cream. Instruct patients to use sunscreen and wear protective clothing (e.g., a hat). Advise patients not to use Imiquimod Cream until fully recovered from a sunburn.
ADVERSE REACTIONS
The following clinically significant adverse reactions are described elsewhere in the labeling:
Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Actinic Keratosis
The data described below reflect exposure to Imiquimod Cream or vehicle in 436 subjects with AK enrolled in two double-blind, vehicle-controlled trials (Studies AK1 and AK2) [see Clinical Studies (14.1)] . Subjects applied Imiquimod Cream, 5% or vehicle topically, to a 25 cm 2 contiguous treatment area on the face or scalp once daily 2 times per week for 16 weeks.
The incidence of selected adverse reactions reported by ≥1% of subjects during the trials is presented in Table 2.
Table 2: Selected Adverse Reactions Occurring in ≥1% of Imiquimod-Treated Subjects with AK and at a Greater Frequency than Vehicle in Studies AK1 and AK2
Imiquimod Cream (n= 215) | Vehicle (n= 221) | |
Application Site Reaction | 71 (33%) | 32 (14%) |
Upper Respiratory Tract Infection | 33 (15%) | 27 (12%) |
Sinusitis | 16 (7%) | 14 (6%) |
Headache | 11 (5%) | 7 (3%) |
Carcinoma Squamous | 8 (4%) | 5 (2%) |
Diarrhea | 6 (3%) | 2 (1%) |
Eczema | 4 (2%) | 3 (1%) |
Back Pain | 3 (1%) | 2 (1%) |
Fatigue | 3 (1%) | 2 (1%) |
Fibrillation Atrial | 3 (1%) | 2 (1%) |
Infection Viral | 3 (1%) | 2 (1%) |
Dizziness | 3 (1%) | 1 (<1%) |
Vomiting | 3 (1%) | 1 (<1%) |
Urinary Tract Infection | 3 (1%) | 1 (<1%) |
Fever | 3 (1%) | 0 (0%) |
Rigors | 3 (1%) | 0 (0%) |
Alopecia | 3 (1%) | 0 (0%) |
The incidence of application site reactions reported by >1% of subjects during the trials is presented in Table 3.
Table 3: Application Site Reactions Reported by >1% of Imiquimod-Treated Subjects with AK and at a Greater Frequency than Vehicle in Studies AK1 and AK2
Imiquimod Cream (n= 215) | Vehicle (n= 221) | |
Itching | 44 (20%) | 17 (8%) |
Burning | 13 (6%) | 4 (2%) |
Bleeding | 7 (3%) | 1 (<1%) |
Stinging | 6 (3%) | 2 (1%) |
Pain | 6 (3%) | 2 (1%) |
Induration | 5 (2%) | 3 (1%) |
Tenderness | 4 (2%) | 3 (1%) |
Irritation | 4 (2%) | 0 (0%) |
Local skin reactions were collected independently of the adverse reaction "application site reaction". The incidence and severity of local skin reactions that occurred during controlled trials are shown in Table 4.
Table 4: Local Skin Reactions in the Treatment Area of Imiquimod-Treated Subjects with AK as Assessed by the Investigator in Studies AK1 and AK2
Imiquimod Cream (n= 215) | Vehicle (n= 220) | |||
All Grades• | Severe | All Grades• | Severe | |
Erythema | 209 (97%) | 38 (18%) | 206 (93%) | 5 (2%) |
Flaking/Scaling/Dryness | 199 (93%) | 16 (7%) | 199 (91%) | 7 (3%) |
Scabbing/Crusting | 169 (79%) | 18 (8%) | 92 (42%) | 4 (2%) |
Edema | 106 (49%) | 0 (0%) | 22 (10%) | 0 (0%) |
Erosion/Ulceration | 103 (48%) | 5 (2%) | 20 (9%) | 0 (0%) |
Weeping/Exudate | 45 (22%) | 0 (0%) | 3 (1%) | 0 (0%) |
Vesicles | 19 (9%) | 0 (0%) | 2 (1%) | 0 (0%) |
•Mild, Moderate, or Severe
The adverse reactions that most frequently resulted in clinical intervention (e.g., rest periods, withdrawal from trial) were local skin and application site reactions. In the trials, 2% (5/215) of subjects discontinued for local skin/application site reactions. Of the 215 subjects treated, 35 subjects (16%) on imiquimod cream and 3 of 220 subjects (1%) on vehicle had at least one rest period. Of the imiquimod-treated subjects, 32 (91%) resumed therapy after a rest period.
In the AK trials, 22 of 678 (3.2%) of imiquimod-treated subjects developed treatment site infections that required a rest period off imiquimod cream and were treated with antibiotics (19 with oral and 3 with topical).
Of the 206 imiquimod-treated subjects with both baseline and 8-week post-treatment scarring assessments, 6 (2.9%) had a greater degree of scarring scores at 8 weeks post-treatment than at baseline.
Superficial Basal Cell Carcinoma
The data described below reflect exposure to imiquimod cream or vehicle in 364 subjects with sBCC enrolled in two double-blind, vehicle-controlled trials (sBCC1 and sBCC2) [see Clinical Studies (14.2 )] . Subjects applied imiquimod cream, 5% or vehicle topically 5 times per week for 6 weeks.
The incidence of selected adverse reactions reported by ≥1% of subjects during the trials is summarized in Table 5.
Table 5: Selected Adverse Reactions Reported by ≥1% of Imiquimod-Treated Subjects with sBCC and at a Greater Frequency than Vehicle in Studies sBCC1 and sBCC2
Imiquimod Cream (n= 185) N % | Vehicle (n= 179) N % | |
Application Site Reaction | 52 (28%) | 5 (3%) |
Headache | 14 (8%) | 4 (2%) |
Back Pain | 7 (4%) | 1 (<1%) |
Upper Respiratory Tract Infection | 6 (3%) | 2 (1%) |
Rhinitis | 5 (3%) | 1 (<1%) |
Lymphadenopathy | 5 (3%) | 1 (<1%) |
Fatigue | 4 (2%) | 2 (1%) |
Sinusitis | 4 (2%) | 1 (<1%) |
Dyspepsia | 3 (2%) | 2 (1%) |
Coughing | 3 (2%) | 1 (<1%) |
Fever | 3 (2%) | 0 (0%) |
Dizziness | 2 (1%) | 1 (<1%) |
Anxiety | 2 (1%) | 1 (<1%) |
Pharyngitis | 2 (1%) | 1 (<1%) |
Chest Pain | 2 (1%) | 0 (0%) |
Nausea | 2 (1%) | 0 (0%) |
The most frequently reported adverse reactions were local skin and application site reactions. The incidence of application site reactions reported by >1% of the subjects during the 6-week treatment period is summarized in Table 6.
Table 6: Application Site Reactions Reported by > 1% of Imiquimod-Treated Subjects with sBCC and at a Greater Frequency than Vehicle in Studies sBCC1 and sBCC2
Imiquimod Cream (n= 185) | Vehicle (n= 179) | |
Itching | 30 (16%) | 1 (1%) |
Burning | 11 (6%) | 2 (1%) |
Pain | 6 (3%) | 0 (0%) |
Bleeding | 4 (2%) | 0 (0%) |
Erythema | 3 (2%) | 0 (0%) |
Papule(s) | 3 (2%) | 0 (0%) |
Tenderness | 2 (1%) | 0 (0%) |
Infection | 2 (1%) | 0 (0%) |
Local skin reactions were collected independently of the adverse reaction “application site reaction”. The incidence and severity of local skin reactions that occurred during the controlled trials are shown in Table 7.
Table 7: Local Skin Reactions in the Treatment Area of Imiquimod-Treated Subjects with sBCC as Assessed by the Investigator in Studies sBCC1 and sBCC2
Imiquimod Cream (n= 184) | Vehicle (n= 178) | |||
All Grades• | Severe | All Grades• | Severe | |
Erythema | 184 (100%) | 57 (31%) | 173 (97%) | 4 (2%) |
Flaking/Scaling | 167 (91%) | 7 (4%) | 135 (76%) | 0 (0%) |
Induration | 154 (84%) | 11 (6%) | 94 (53%) | 0 (0%) |
Scabbing/Crusting | 152 (83%) | 35 (19%) | 61 (34%) | 0 (0%) |
Edema | 143 (78%) | 13 (7%) | 64 (36%) | 0 (0%) |
Erosion | 122 (66%) | 23 (13%) | 25 (14%) | 0 (0%) |
Ulceration | 73 (40%) | 11 (6%) | 6 (3%) | 0 (0%) |
Vesicles | 57 (31%) | 3 (2%) | 4 (2%) | 0 (0%) |
•Mild, Moderate, or Severe
The adverse reactions that most frequently resulted in clinical intervention (e.g., rest periods, withdrawal from trial) were local skin and application site reactions; 10% (19/185) of imiquimod-treated subjects received rest periods. The average number of doses not received per imiquimod-treated subject due to rest periods was 7 doses with a range of 2 to 22 doses; 79% of subjects (15/19) resumed therapy after a rest period. Overall, in the clinical trials, 2% (4/185) of imiquimod-treated subjects discontinued for local skin/application site reactions.
In the sBCC trials, 17 of 1266 (1.3%) imiquimod-treated subjects developed treatment site infections that required a rest period and treatment with antibiotics.
External Genital Warts
In controlled clinical trials for EGW, including a double-blind, vehicle-controlled clinical trial in 209 adult subjects with EGW (Study EGW1) [see Clinical Studies (14.3 )] , imiquimod cream, 5% was applied topically to EGW in 109 subjects. Selected adverse reactions in imiquimod-treated subjects are listed below (see Table 8).
Table 8: Selected Adverse Reactions in Imiquimod-Treated Subjects with EGW in Vehicle-Controlled Clinical Trials
Females | Males | |||
Imiquimod Cream (n=117) | Vehicle (n=103) | Imiquimod Cream (n=156) | Vehicle (n=158) | |
Wart Site | ||||
Itching | 38 (32%) | 21 (20%) | 34 (22%) | 16 (10%) |
Burning | 30 (26%) | 12 (12%) | 14 (9%) | 8 (5%) |
Pain | 9 (8%) | 2 (2%) | 3 (2%) | 1 (1%) |
Soreness | 3 (3%) | 0 (0%) | 0 (0%) | 1 (1%) |
Fungal Infection | 13 (11%) | 3 (3%) | 3 (2%) | 1 (1%) |
Systemic Reactions | ||||
Headache | 5 (4%) | 3 (3%) | 8 (5%) | 3 (2%) |
Influenza-like Symptoms | 4 (3%) | 2 (2%) | 2 (1%) | 0 (0%) |
Myalgia | 1 (1%) | 0 (0%) | 2 (1%) | 1 (1%) |
The most frequently reported adverse reactions were local skin and application site reactions.
Overall, 1.2% (4/327) of the subjects discontinued treatment due to local skin/application site reactions.
The incidence and severity of local skin reactions during controlled clinical trials are shown in Table 9.
Table 9: Local Skin Reactions in the Treatment Area of Imiquimod-Treated Subjects with EGW as Assessed by the Investigator in Vehicle-Controlled Clinical Trials
Imiquimod Cream | Vehicle | |||||||
Females (n=114) | Males (n=156) | Females (n=99) | Males (n=157) | |||||
All Grades• | Severe | All Grades• | Severe | All Grades• | Severe | All Grades• | Severe | |
Erythema | 74 (65%) | 4 (4%) | 90 (58%) | 6 (4%) | 21 (21%) | 0 (0%) | 34 (22%) | 0 (0%) |
Erosion | 35 (31%) | 1 (1%) | 47 (30%) | 2 (1%) | 8 (8%) | 0 (0%) | 10 (6%) | 0 (0%) |
Excoriation/ Flaking | 21 (18%) | 0 (0%) | 40 (26%) | 1 (1%) | 8 (8%) | 0 (0%) | 12 (8%) | 0 (0%) |
Edema | 20 (18%) | 1 (1%) | 19 (12%) | 0 (0%) | 5 (5%) | 0 (0%) | 1 (1%) | 0 (0%) |
Scabbing | 4 (4%) | 0 (0%) | 20 (13%) | 0 (0%) | 0 (0%) | 0 (0%) | 4 (3%) | 0 (0%) |
Induration | 6 (5%) | 0 (0%) | 11 (7%) | 0 (0%) | 2 (2%) | 0 (0%) | 3 (2%) | 0 (0%) |
Ulceration | 9 (8%) | 3 (3%) | 7 (4%) | 0 (0%) | 1 (1%) | 0 (0%) | 1 (1%) | 0 (0%) |
Vesicles | 3 (3%) | 0 (0%) | 3 (2%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) |
•Mild, Moderate, or Severe
Remote site skin reactions were also reported. The severe remote site skin reactions reported for females were erythema (3%), ulceration (2%), and edema (1%); and for males, erosion (2%), and erythema, edema, induration, and excoriation/flaking (each 1%).
Other adverse reactions reported by more than 1% of imiquimod-treated subjects included:
Application Site Disorders: hypopigmentation, irritation, rash, sensitivity, stinging, tenderness
Body as a Whole: fatigue, fever
Gastrointestinal System Disorders: diarrhea
Remote Site Reactions: bleeding, burning, itching, pain, tenderness, tinea cruris
Postmarketing Experience
The following adverse reactions have been identified during post-approval use of imiquimod cream. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Application Site Disorders: tingling at the application site
Body as a Whole: angioedema
Cardiovascular: capillary leak syndrome, cardiac failure, cardiomyopathy, pulmonary edema, arrhythmias (tachycardia, atrial fibrillation, palpitations), chest pain, ischemia, myocardial infarction, syncope
Endocrine: thyroiditis
Gastrointestinal System Disorders: abdominal pain
Hematological: decreases in red cell, white cell, and platelet counts (including idiopathic thrombocytopenic purpura), lymphoma
Hepatic: abnormal liver function
Infections and Infestations: herpes simplex
Musculoskeletal System Disorders: arthralgia
Neuropsychiatric: agitation, cerebrovascular accident, convulsions (including febrile convulsions), depression, insomnia, multiple sclerosis aggravation, paresis, suicide
Respiratory: dyspnea
Urinary System Disorders: proteinuria, dysuria, urinary retention
Skin and Appendages: exfoliative dermatitis, erythema multiforme, hypertrophic scar, hyperpigmentation, hypopigmentation, including complete depigmentation.
Vascular: Henoch-Schönlein purpura syndrome
DESCRIPTION
Imiquimod Cream, 5% is an immune response modifier for topical administration. Each gram contains 50 mg of imiquimod in an off-white oil-in-water vanishing cream base consisting of benzyl alcohol, cetyl alcohol, glycerin, methylparaben, oleic acid, oleyl alcohol, polysorbate 60, propylparaben, purified water, stearyl alcohol, sorbitan monostearate, white petrolatum, and xanthan gum.
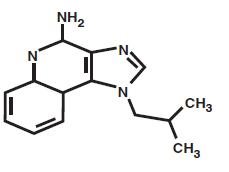
Chemically, imiquimod is 1-(2-methylpropyl)-1 H -imidazo[4,5-c]quinolin-4-amine. Imiquimod has a molecular formula of C 14 H 16 N 4 and a molecular weight of 240.3. Its structural formula is:
CLINICAL PHARMACOLOGY
Mechanism of Action
The mechanism of action of Imiquimod Cream in treating AK, sBCC, and EGW lesions is unknown.
Pharmacodynamics
Actinic Keratosis
In a trial of 18 subjects with AK comparing imiquimod cream to vehicle, increases from baseline in Week 2 biomarker levels were reported for CD3, CD4, CD8, CD11c, and CD68 for imiquimod-treated subjects; however, the clinical relevance of these findings is unknown.
Superficial Basal Cell Carcinoma
An open-label trial in 6 subjects with sBCC suggests that treatment with imiquimod cream may increase the infiltration of lymphocytes, dendritic cells, and macrophages into the tumor lesion; however, the clinical significance of these findings is unknown.
External Genital Warts
Imiquimod has no direct antiviral activity in cell culture. A trial in 22 subjects with EGW comparing imiquimod cream and vehicle shows that imiquimod cream induces mRNA encoding cytokines including interferon-ɑ at the treatment site. In addition, HPVL1 mRNA and HPV DNA are significantly decreased following treatment. However, the clinical relevance of these findings is unknown.
Pharmacokinetics
Absorption
Systemic absorption of imiquimod across the affected skin of 58 subjects with AK was observed with a dosing frequency of 3 applications per week for 16 weeks. Mean peak serum drug concentrations at the end of Week 16 were approximately 0.1, 0.2, and 3.5 ng/mL for the applications to face (12.5 mg imiquimod, 1 unit-dose packet), scalp (25 mg, 2 packets), and hands/arms (75 mg, 6 packets), respectively.
Table 10: Mean Serum Imiquimod Concentration in Adults Following Administration of the Last Topical Dose during Week 16 (Actinic Keratosis)
Amount of Imiquimod Cream Applied | Mean Peak Serum Imiquimod Concentration [C max ] |
12.5 mg (1 packet) | 0.1 ng/mL |
25 mg (2 packets) | 0.2 ng/mL |
75 mg (6 packets) | 3.5 ng/mL |
The application surface area was not controlled when more than 1 packet was used. Dose proportionality was not observed. However, it appears that systemic exposure may be more dependent on surface area of application than amount of applied dose. The apparent half-life was approximately 10 times greater with topical dosing than the 2-hour apparent half-life seen following subcutaneous dosing, suggesting prolonged retention of drug in the skin. Mean urinary recoveries of imiquimod and metabolites combined were 0.08% and 0.15% of the applied dose in the group using 75 mg (6 packets) for males and females, respectively following 3 applications per week for 16 weeks.
Systemic absorption of imiquimod was observed across the affected skin of 12 subjects with genital/perianal warts, with an average dose of 4.6 mg. Mean peak drug concentration of approximately 0.4 ng/mL was seen during the trial. Mean urinary recoveries of imiquimod and metabolites combined over the whole course of treatment, expressed as percent of the estimated applied dose, were 0.11% and 2.41% in the males and females, respectively.
NONCLINICAL TOXICOLOGY
Carcinogenesis, Mutagenesis, Impairment of Fertility
In an oral (gavage) rat carcinogenicity study, imiquimod was administered to Wistar rats on a 2 times per week (up to 6 mg/kg/day) or daily (3 mg/kg/day) dosing schedule for 24 months. No treatment-related tumors were noted in the oral rat carcinogenicity study up to the highest doses tested in this study of 6 mg/kg administered 2 times per week in female rats (87 times the MRHD based on weekly AUC comparison), 4 mg/kg administered 2 times per week in male rats (75 times the MRHD based on weekly AUC comparison), or 3 mg/kg administered 7 times per week to male and female rats (153 times the MRHD based on weekly AUC comparison).
In a dermal mouse carcinogenicity study, imiquimod cream (up to 5 mg/kg/application imiquimod or 0.3% imiquimod cream) was applied to the backs of mice 3 times per week for 24 months. A statistically significant increase in the incidence of liver adenomas and carcinomas was noted in high dose male mice compared to control male mice (251 times the MRHD based on weekly AUC comparison). An increased number of skin papillomas was observed in vehicle cream control group animals at the treated site only.
Imiquimod revealed no evidence of mutagenic or clastogenic potential based on the results of five in vitro genotoxicity tests (Ames assay, mouse lymphoma L5178Y assay, Chinese hamster ovary cell chromosome aberration assay, human lymphocyte chromosome aberration assay, and SHE cell transformation assay) and three in vivo genotoxicity tests (rat and hamster bone marrow cytogenetics assay and a mouse-dominant lethal test). Daily oral administration of imiquimod to rats, throughout mating, gestation, parturition, and lactation, demonstrated no effects on growth, fertility or reproduction, at doses up to 87 times the MRHD based on AUC comparison.
CLINICAL STUDIES
Actinic Keratosis
In two double-blind, vehicle-controlled clinical trials, 436 subjects with AK were randomized to treatment with either imiquimod cream, 5% or vehicle applied topically once daily 2 times per week for 16 weeks (Studies AK1 and AK2). The trials enrolled subjects with 4 to 8 clinically typical, visible, discrete, nonhyperkeratotic, nonhypertrophic AK lesions within a 25 cm 2 contiguous treatment area on either the face or scalp. The 25 cm 2 contiguous treatment area could be of any dimensions (e.g., 5 cm × 5 cm, 3 cm × 8.3 cm, 2 cm × 12.5 cm). Trial subjects ranged from 37 to 88 years of age (median 66 years) and 55% had Fitzpatrick skin type I or II. All imiquimod-treated subjects were White.
On a scheduled dosing day, the trial cream was applied to the entire treatment area prior to normal sleeping hours and left on for approximately 8 hours. Twice-weekly dosing was continued for a total of 16 weeks. The clinical response of each subject was evaluated 8 weeks after the last scheduled application of trial cream. Efficacy was assessed by the complete clearance rate, defined as the proportion of subjects at the 8-week post-treatment visit with no (zero) clinically visible AK lesions in the treatment area. Complete clearance included clearance of all baseline lesions, as well as any new or subclinical AK lesions which appeared during therapy.
Complete and partial clearance rates are shown in Table 11. The partial clearance rate was defined as the percentage of subjects in whom 75% or more baseline AK lesions were cleared.
Table 11: Clearance Rates of AK at 8 Weeks Post-Treatment
Complete Clearance Rates (100% AK Lesions Cleared) | ||
Trial | Imiquimod Cream | Vehicle |
Study AK1 | 46% (49/107) | 3% (3/110) |
Study AK2 | 44% (48/108) | 4% (4/111) |
Partial and Complete Clearance Rates (75% or More Baseline AK Lesions Cleared) | ||
Trial | Imiquimod Cream | Vehicle |
Study AK1 | 60% (64/107) | 10% (11/110) |
Study AK2 | 58% (63/108) | 14% (15/111) |
During treatment, 48% (103/215) of imiquimod-treated subjects experienced an increase in AK lesions relative to the number present at baseline within the treatment area. Subjects with an increase in AK lesions had a similar response to those with no increase in AK lesions.
Superficial Basal Cell Carcinoma
In two double-blind, vehicle-controlled clinical trials, 364 subjects with primary sBCC were treated with imiquimod cream, 5% or vehicle applied topically once daily 5 times per week for 6 weeks (Studies sBCC1 and sBCC2). Target tumors were biopsy-confirmed sBCC and had a minimum area of 0.5 cm 2 and a maximum diameter of 2.0 cm (4.0 cm 2 ). Target tumors were not to be located within 1.0 cm of the hairline, or on the anogenital area or on the hands or feet, or to have any atypical features. The population ranged from 31 to 89 years of age (median 60 years) and 65% had Fitzpatrick skin type I or II. On a scheduled dosing day, trial cream was applied to the target tumor and approximately 1 cm (about 1/3 inch) beyond the target tumor prior to normal sleeping hours, and 5 times per week dosing was continued for a total of 6 weeks. The target tumor area was clinically assessed 12 weeks after the last scheduled application of trial cream. The entire target tumor was then excised and examined histologically for the presence of tumor.
Efficacy was assessed by the complete response rate defined as the proportion of subjects with clinical (visual) and histological clearance of the sBCC lesion at 12 weeks post-treatment. Of imiquimod-treated subjects, 6% (11/178) who had both clinical and histological assessments post-treatment, and who appeared to be clinically clear had evidence of tumor on excision of the clinically clear treatment area.
Data on composite clearance (defined as both clinical and histological clearance) are shown in Table 12.
Table 12: Composite Clearance Rates at 12 Weeks Post-Treatment for sBCC
Trial | Imiquimod Cream | Vehicle |
Study sBCC1 | 70% (66/94) | 2% (2/89) |
Study sBCC2 | 80% (73/91) | 1% (1/90) |
Total | 75% (139/185) | 2% (3/179) |
A separate 5-year, open-label trial was conducted to assess the recurrence of sBCC treated with imiquimod cream applied topically once daily 5 days per week for 6 weeks. Target tumor inclusion criteria were the same as for the trials described above. At 12 weeks post-treatment, subjects were clinically evaluated for evidence of persistent sBCC (no histological assessment). Subjects with no clinical evidence of sBCC entered the long-term follow-up period. At the 12-week post-treatment assessment, 90% (163/182) of the subjects enrolled had no clinical evidence of sBCC at their target site and 162 subjects entered the long-term follow-up period for up to 5 years. Two-year (24-month) follow-up data are available from this trial and are presented in Table 13.
Table 13: Estimated Clinical Clearance Rates for sBCC in Imiquimod-Treated Subjects During Follow-up Period in Open-Label Trial
Follow-up Visit after 12-Week Post-Treatment Assessment | No. of Subjects Who Remained Clinically Clear | No. of Subjects with sBCC Recurrence | No. of Subjects Who Discontinued at This Visit with No sBCC a | Estimated Rate of Subjects Who Clinically Cleared and Remained Clear b |
Month 3 | 153 | 4 | 5 | 87% |
Month 6 | 149 | 4 | 0 | 85% |
Month 12 | 143 | 2 | 4 | 84% |
Month 24 | 139 | 4 | 0 | 79% |
a Reasons for discontinuation included death, noncompliance, entry criteria violations, personal reasons, and treatment of nearby sBCC tumor.
b Estimated rate of subjects who clinically cleared and remained clear are estimated based on the time to event analysis employing the life table method beginning with the rate of clinical clearance at 12 weeks post-treatment.
External Genital Warts
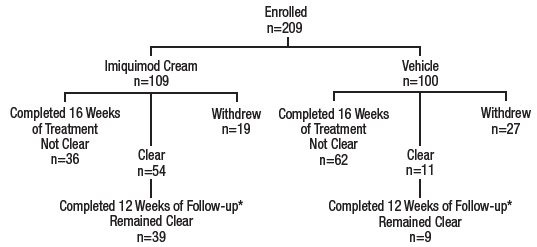
In a double-blind, placebo-controlled clinical trial, 209 otherwise healthy subjects 18 years and older with EGW were treated with imiquimod cream, 5% or vehicle applied topically once daily 3 times per week for a maximum of 16 weeks (Study EGW1). The median baseline wart area was 69 mm 2 (range 8 to 5525 mm 2 ). Subject accountability is shown in the figure below.
Figure 1: Subject Accountability for Study EGW1
 • The other subjects were either lost to follow-up or experienced recurrences.
• The other subjects were either lost to follow-up or experienced recurrences.
Data on complete clearance are listed in Table 14. The median time to complete wart clearance was 10 weeks.
Table 14: Complete Clearance Rates of EGW in Study EGW1
Treatment | Subjects with Complete Clearance of Warts | Subjects Without Follow-up | Subjects with Warts Remaining at Week 16 |
Overall | |||
Imiquimod Cream (n =109) | 54 (50%) | 19 (17%) | 36 (33%) |
Vehicle (n =100) | 11 (11%) | 27 (27%) | 62 (62%) |
Females | |||
Imiquimod Cream (n =46) | 33 (72%) | 5 (11%) | 8 (17%) |
Vehicle (n =40) | 8 (20%) | 13 (33%) | 19 (48%) |
Males | |||
Imiquimod Cream (n =63) | 21 (33%) | 14 (22%) | 28 (44%) |
Vehicle (n =60) | 3 (5%) | 14 (23%) | 43 (72%) |
HOW SUPPLIED/STORAGE AND HANDLING
Imiquimod Cream, 5% is supplied in unit-dose packets each of which contains 250 mg of a white to off-white cream.
Available as: a box of 12 packets (NDC 45802- 368 -53) and a box of 24 packets (NDC 45802- 368 -62).
Store at 4°-25°C (39°-77°F). Avoid freezing.
Mechanism of Action
The mechanism of action of Imiquimod Cream in treating AK, sBCC, and EGW lesions is unknown.