Get your patient on Lumryz - Sodium Oxybate for Suspension, Extended Release (Sodium Oxybate)
Lumryz - Sodium Oxybate for Suspension, Extended Release prescribing information
WARNING: CENTRAL NERVOUS SYSTEM (CNS) DEPRESSION AND ABUSE AND MISUSE
Central Nervous System Depression
LUMRYZ (sodium oxybate) is a CNS depressant. Clinically significant respiratory depression and obtundation may occur in patients treated with LUMRYZ at recommended doses [see Warnings and Precautions (5.1) ] . Many patients who received sodium oxybate during clinical trials in narcolepsy were receiving central nervous system stimulants [see Clinical Trials (14) ] .
Abuse and Misuse
LUMRYZ (sodium oxybate) is the sodium salt of gamma-hydroxybutyrate (GHB). Abuse or misuse of illicit GHB, either alone or in combination with other CNS depressants, is associated with CNS adverse reactions, including seizure, respiratory depression, decreases in the level of consciousness, coma, and death [see Warnings and Precautions (5.2) ] .
Because of the risks of CNS depression and abuse and misuse, LUMRYZ is available only through a restricted program under a Risk Evaluation and Mitigation Strategy (REMS) called the LUMRYZ REMS [see Warnings and Precautions (5.3) ].
1 INDICATIONS AND USAGE
LUMRYZ is indicated for the treatment of cataplexy or excessive daytime sleepiness (EDS) in patients 7 years of age and older with narcolepsy.
2 DOSAGE AND ADMINISTRATION
Adult Dosing Information
The recommended starting dosage of LUMRYZ in adults is 4.5 grams (g) once per night administered orally. Increase the dosage by 1.5 g per night at weekly intervals to the recommended dosage range of 6 g to 9 g once per night orally. The dosage may be gradually titrated based on efficacy and tolerability. Doses higher than 9 g per night have not been studied and should not ordinarily be administered.
Pediatric Dosing Information
The recommended starting pediatric dosage, titration regimen, and maximum total nightly dosage are based on patient weight.
Pediatric Patients 7 years and Older Weighing at least 45 kg
The recommended starting dosage of LUMRYZ in pediatric patients 7 years and older weighing at least 45 kg is 4.5 g once per night administered orally. Increase the dosage by 1.5 g per night at weekly intervals to the maximum recommended dosage of 9 g once per night orally. The dosage may be gradually titrated based on efficacy and tolerability.
Pediatric Patients 7 years and Older Weighing Less than 45 kg
Because the recommended starting dosage in pediatric patients 7 years and older weighing less than 45 kg cannot be achieved with the available strengths of LUMRYZ, use another sodium oxybate product to initiate treatment. Refer to the Prescribing Information of other sodium oxybate products for the recommended dosage for those products.
The maximum recommended dosage for patients 7 years and older weighing 20 kg to <30 kg is 6 g once per night orally, and the maximum recommended dosage for patients 7 years and older weighing 30 kg to <45 kg is 7.5 g once per night orally [ see Dosage and Administration (2.4 ) ]. There is insufficient information to provide specific dosing recommendations for patients 7 years and older who weigh less than 20 kg.
Important Administration Instructions
LUMRYZ is taken orally as a single dose at bedtime. Prepare the dose of LUMRYZ prior to bedtime. Prior to ingestion, the dose of LUMRYZ should be suspended in approximately 1/3 cup (approximately 80 mL) of water (with or without calorie-free drink mix or flavored water enhancer) in the mixing cup provided [see Instructions for Use ] . Do not use hot water [see Clinical Pharmacology (12.3) ] . After mixing, consume LUMRYZ within 30 minutes.
Take LUMRYZ at least 2 hours after eating [see Clinical Pharmacology (12.3) ].
Patients should take LUMRYZ while in bed and lie down immediately after dosing as LUMRYZ may cause them to fall asleep abruptly without first feeling drowsy. Patients will often fall asleep within 5 minutes of taking LUMRYZ, and will usually fall asleep within 15 minutes, though the time it takes any individual patient to fall asleep may vary from night to night. Patients should remain in bed following ingestion of LUMRYZ.
Switching Patients from Immediate-Release Sodium Oxybate
Patients who are currently being treated with immediate-release sodium oxybate may be switched to LUMRYZ at the nearest equivalent dosage in grams per night (e.g., 7.5 g sodium oxybate divided into two 3.75 g doses per night to 7.5 g LUMRYZ once per night).
3 DOSAGE FORMS AND STRENGTHS
For extended-release oral suspension: LUMRYZ is a white to off-white powder provided in packets of 4.5 g, 6 g, 7.5 g, or 9 g of sodium oxybate.
8 USE IN SPECIFIC POPULATIONS
Pregnancy
Risk Summary
There are no adequate data on the developmental risk associated with the use of sodium oxybate in pregnant women. Oral administration of sodium oxybate to pregnant rats (150, 350, or 1,000 mg/kg/day) or rabbits (300, 600, or 1,200 mg/kg/day) throughout organogenesis produced no clear evidence of developmental toxicity; however, oral administration to rats throughout pregnancy and lactation resulted in increased stillbirths and decreased offspring postnatal viability and growth, at a clinically relevant dose [see Data] .
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively. The background risk of major birth defects and miscarriage for the indicated population is unknown.
Clinical Considerations
Labor or Delivery
LUMRYZ has not been studied in labor or delivery. In obstetric anesthesia using an injectable formulation of sodium oxybate, newborns had stable cardiovascular and respiratory measures but were very sleepy, causing a slight decrease in Apgar scores. There was a fall in the rate of uterine contractions 20 minutes after injection. Placental transfer is rapid and gamma-hydroxybutyrate (GHB) has been detected in newborns at delivery after intravenous administration of GHB to mothers. Subsequent effects of sodium oxybate on later growth, development, and maturation in humans are unknown.
Data
Animal Data
Oral administration of sodium oxybate to pregnant rats (150, 350, or 1,000 mg/kg/day) or rabbits (300, 600, or 1,200 mg/kg/day) throughout organogenesis produced no clear evidence of developmental toxicity. The highest doses tested in rats and rabbits were approximately 1 and 3 times, respectively, the maximum recommended human dose (MRHD) of 9 g per night on a body surface area (mg/m 2 ) basis.
Oral administration of sodium oxybate (150, 350, or 1,000 mg/kg/day) to rats throughout pregnancy and lactation resulted in increased stillbirths and decreased offspring postnatal viability and body weight gain at the highest dose tested. The no-effect dose for pre- and postnatal developmental toxicity in rats is less than the MRHD on a mg/m 2 basis.
Lactation
Risk Summary
GHB is excreted in human milk after oral administration of sodium oxybate. There is insufficient information on the risk to a breastfed infant, and there is insufficient information on milk production in nursing mothers. The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for LUMRYZ and any potential adverse effects on the breastfed infant from LUMRYZ or from the underlying maternal condition.
Pediatric Use
LUMRYZ has not been studied in a pediatric clinical trial. The safety and effectiveness of LUMRYZ in the treatment of cataplexy or excessive daytime sleepiness in pediatric patients 7 years of age and older with narcolepsy is supported by evidence from a double-blind, placebo-controlled, randomized-withdrawal study of immediate-release sodium oxybate [see Adverse Reactions (6.1) and Clinical Studies (14.2) ].
In the pediatric clinical trial with immediate-release sodium oxybate administration in pediatric patients 7 years of age and older with narcolepsy, serious adverse reactions of central sleep apnea and oxygen desaturation documented by polysomnography evaluation; depression; suicidal ideation; neuropsychiatric reactions including acute psychosis, confusion, and anxiety; and parasomnias, including sleepwalking, have been reported [see Warnings and Precautions (5.4 , 5.5 , 5.6 , 5.7 ) and Adverse Reactions (6.1) ].
The safety and effectiveness of LUMRYZ have not been established in pediatric patients younger than 7 years old.
Juvenile Animal Toxicity Data
In a study in which sodium oxybate (0, 100, 300, or 900 mg/kg/day) was orally administered to rats during the juvenile period of development (postnatal days 21 through 90), mortality was observed at the two highest doses tested. Deaths occurred during the first week of dosing and were associated with clinical signs (including decreased activity and respiratory rate) consistent with the pharmacological effects of the drug. Reduced body weight gain in males and females and delayed sexual maturation in males were observed at the highest dose tested. The no-effect dose for adverse effects in juvenile rats is associated with plasma exposures (AUC) less than that at the maximum recommended human dose (9 g/night).
Geriatric Use
Clinical studies of LUMRYZ or immediate-release sodium oxybate in patients with narcolepsy did not include sufficient numbers of subjects age 65 years and older to determine whether they respond differently from younger subjects. In controlled trials of immediate-release sodium oxybate in another population, 39 (5%) of 874 patients were 65 years or older. Discontinuations of treatment due to adverse reactions were increased in the elderly compared to younger adults (21% vs. 19%). Frequency of headaches was markedly increased in the elderly (39% vs. 19%). The most common adverse reactions were similar in both age categories. In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
Hepatic Impairment
Because of an increase in exposure to LUMRYZ, LUMRYZ should not be initiated in patients with hepatic impairment because appropriate dosage adjustments for initiation of LUMRYZ cannot be made with the available dosage strengths [see Clinical Pharmacology (12.3) ] . Patients with hepatic impairment who have been titrated to a maintenance dosage of another oxybate product can be switched to LUMRYZ if the appropriate dosage strength is available.
4 CONTRAINDICATIONS
LUMRYZ is contraindicated for use in:
● combination with sedative hypnotics [see Warnings and Precautions (5.1) ]
● combination with alcohol [see Warnings and Precautions (5.1) ]
● patients with succinic semialdehyde dehydrogenase deficiency [see Clinical Pharmacology (12.3) ]
5 WARNINGS AND PRECAUTIONS
Central Nervous System Depression
LUMRYZ is a central nervous system (CNS) depressant. Clinically significant respiratory depression and obtundation has occurred in patients treated with immediate-release sodium oxybate at recommended doses in clinical trials and may occur in patients treated with LUMRYZ at recommended doses. LUMRYZ is contraindicated in combination with alcohol and sedative hypnotics. The concurrent use of LUMRYZ with other CNS depressants, including but not limited to opioid analgesics, benzodiazepines, sedating antidepressants or antipsychotics, sedating antiepileptic drugs, general anesthetics, muscle relaxants, and/or illicit CNS depressants, may increase the risk of respiratory depression, hypotension, profound sedation, syncope, and death. If use of these CNS depressants in combination with LUMRYZ is required, dose reduction or discontinuation of one or more CNS depressants (including LUMRYZ) should be considered. In addition, if short-term use of an opioid (e.g., post- or perioperative) is required, interruption of treatment with LUMRYZ should be considered. In addition to coadministration of LUMRYZ and alcohol being contraindicated because of respiratory depression, consumption of alcohol while taking LUMRYZ may also result in a more rapid release of the dose of sodium oxybate [see Clinical Pharmacology (12.3) ] .
Healthcare providers should caution patients about operating hazardous machinery, including automobiles or airplanes, until they are reasonably certain that LUMRYZ does not affect them adversely (e.g., impair judgment, thinking, or motor skills). Patients should not engage in hazardous occupations or activities requiring complete mental alertness or motor coordination, such as operating machinery or a motor vehicle or flying an airplane, for at least 6 hours after taking LUMRYZ. Patients should be queried about CNS depression-related events upon initiation of LUMRYZ therapy and periodically thereafter.
LUMRYZ is available only through a restricted program under a REMS [see Warnings and Precautions (5.3) ].
Abuse and Misuse
LUMRYZ is a Schedule III controlled substance. The active ingredient of LUMRYZ, sodium oxybate, is the sodium salt of gamma-hydroxybutyrate (GHB), a Schedule I controlled substance. Abuse of illicit GHB, either alone or in combination with other CNS depressants, is associated with CNS adverse reactions, including seizure, respiratory depression, decreases in the level of consciousness, coma, and death. The rapid onset of sedation, coupled with the amnestic features of GHB, particularly when combined with alcohol, has proven to be dangerous for the voluntary and involuntary user (e.g., assault victim). Because illicit use and abuse of GHB have been reported, physicians should carefully evaluate patients for a history of drug abuse and follow such patients closely, observing them for signs of misuse or abuse of GHB (e.g., increase in size or frequency of dosing, drug-seeking behavior, feigned cataplexy) [see Warnings and Precautions (5.3) and Drug Abuse and Dependence (9.2) ].
LUMRYZ is available only through a restricted program under a REMS [see Warnings and Precautions (5.3) ].
LUMRYZ REMS
LUMRYZ is available only through a restricted distribution program called the LUMRYZ REMS because of the risks of central nervous system depression and abuse and misuse [see Warnings and Precautions (5.1 , 5.2) ] .
Notable requirements of the LUMRYZ REMS include the following:
● Healthcare providers who prescribe LUMRYZ are specially certified.
● LUMRYZ will be dispensed only by pharmacies that are specially certified.
● LUMRYZ will be dispensed and shipped only to patients who are enrolled in the LUMRYZ REMS with documentation of safe use conditions.
Further information is available at www.LUMRYZREMS.com or by calling 1-877-453-1029.
Respiratory Depression and Sleep-Disordered Breathing
LUMRYZ may impair respiratory drive, especially in patients with compromised respiratory function. In overdoses of oxybate and with illicit use of GHB, life-threatening respiratory depression has been reported [see Overdosage (10) ] .
Increased apnea and reduced oxygenation may occur with LUMRYZ administration. A significant increase in the number of central apneas and clinically significant oxygen desaturation may occur in patients with obstructive sleep apnea treated with LUMRYZ.
During polysomnographic evaluation (PSG), central sleep apnea and oxygen desaturation were observed in pediatric patients with narcolepsy treated with immediate-release sodium oxybate.
In adult clinical trials of LUMRYZ in patients with narcolepsy, no subjects with apnea/hypopnea indexes greater than 15 were allowed to enroll.
In an adult study assessing the respiratory-depressant effects of immediate-release sodium oxybate at doses up to 9 g per night in 21 patients with narcolepsy, no dose-related changes in oxygen saturation were demonstrated in the group as a whole. One of four patients with preexisting moderate-to-severe sleep apnea had significant worsening of the apnea/hypopnea index during treatment.
In an adult study assessing the effects of immediate-release sodium oxybate 9 g per night in 50 patients with obstructive sleep apnea, immediate-release sodium oxybate did not increase the severity of sleep-disordered breathing and did not adversely affect the average duration and severity of oxygen desaturation overall. However, there was a significant increase in the number of central apneas in patients taking immediate-release sodium oxybate, and clinically significant oxygen desaturation (≤55%) was measured in three patients (6%) after administration, with one patient withdrawing from the study and two continuing after single brief instances of desaturation.
In adult clinical trials in 128 patients with narcolepsy administered immediate-release sodium oxybate, two subjects had profound CNS depression, which resolved after supportive respiratory intervention. Two other patients discontinued immediate-release sodium oxybate because of severe difficulty breathing and an increase in obstructive sleep apnea. In two controlled trials assessing polysomnographic (PSG) measures in adult patients with narcolepsy administered immediate-release sodium oxybate, 40 of 477 patients were included with a baseline apnea/hypopnea index of 16 to 67 events per hour, indicative of mild to severe sleep-disordered breathing. None of the 40 patients had a clinically significant worsening of respiratory function, as measured by apnea/hypopnea index and pulse oximetry at doses of 4.5 g to 9 g per night.
Prescribers should be aware that sleep-related breathing disorders tend to be more prevalent in obese patients, in men, in postmenopausal women not on hormone replacement therapy, and among patients with narcolepsy. Increased central apneas and clinically relevant desaturation events have been observed with immediate-release sodium oxybate administration in adult and pediatric patients.
Depression and Suicidality
Depression, and suicidal ideation and behavior, can occur in patients treated with LUMRYZ.
In an adult clinical trial in patients with narcolepsy administered LUMRYZ [see Adverse Reactions (6.1) ] , there were no suicide attempts, but one patient developed suicidal ideation at the 9 g dose. In adult clinical trials in patients with narcolepsy (n=781) administered immediate-release sodium oxybate, there were two suicides and two attempted suicides in patients treated with immediate-release sodium oxybate, including three patients with a previous history of depressive psychiatric disorder. Of the two suicides, one patient used immediate-release sodium oxybate in conjunction with other drugs. Immediate-release sodium oxybate was not involved in the second suicide. Adverse reactions of depression were reported by 7% of 781 patients treated with immediate-release sodium oxybate, with four patients (<1%) discontinuing because of depression. In most cases, no change in immediate-release sodium oxybate treatment was required.
In a controlled trial in adults with narcolepsy administered LUMRYZ where patients were titrated from 4.5 g to 9 g per night, the incidences of depression were 0% at 4.5 g, 1% at 6 g, 1.1% at 7.5 g, and 1.3% at 9 g. In a controlled adult trial, with patients randomized to fixed doses of 3 g, 6 g, or 9 g per night immediate-release sodium oxybate or placebo, there was a single event of depression at the 3 g per night dose. In another adult controlled trial, with patients titrated from an initial 4.5 g per night starting dose of immediate-release sodium oxybate, the incidences of depression were 1.7%, 1.5%, 3.2%, and 3.6% for the placebo, 4.5 g, 6 g, and 9 g per night doses, respectively.
In a clinical trial in pediatric patients with narcolepsy (n=104) administered immediate-release sodium oxybate, one patient experienced suicidal ideation and two patients reported depression while taking immediate-release sodium oxybate.
The emergence of depression in patients treated with LUMRYZ requires careful and immediate evaluation. Patients with a previous history of a depressive illness and/or suicide attempt should be monitored carefully for the emergence of depressive symptoms while taking LUMRYZ.
Other Behavioral or Psychiatric Adverse Reactions
Other behavioral and psychiatric adverse reactions can occur in patients taking LUMRYZ.
During adult clinical trials in patients with narcolepsy administered LUMRYZ, 2% of 107 patients treated with LUMRYZ experienced a confusional state. During adult clinical trials in patients with narcolepsy administered immediate-release sodium oxybate, 3% of 781 patients treated with immediate-release sodium oxybate experienced confusion, with incidence generally increasing with dose.
No patients treated with LUMRYZ discontinued treatment because of confusion. Less than 1% of patients discontinued the immediate-release sodium oxybate because of confusion. Confusion was reported at all recommended doses of immediate-release sodium oxybate from 6 g to 9 g per night. In a controlled trial in adults where patients were randomized to immediate-release sodium oxybate in fixed total daily doses of 3 g, 6 g, or 9 g per night or placebo, a dose-response relationship for confusion was demonstrated, with 17% of patients at 9 g per night experiencing confusion. In that controlled trial, the confusion resolved in all cases soon after termination of treatment. In one trial where immediate-release sodium oxybate was titrated from an initial 4.5 g per night dose, there was a single event of confusion in one patient at the 9 g per night dose. In the majority of cases in all adult clinical trials in patients with narcolepsy administered immediate-release sodium oxybate, confusion resolved either soon after termination of dosing or with continued treatment.
Anxiety occurred in 7.5% of 107 patients treated with LUMRYZ in the adult trial in patients with narcolepsy. Anxiety occurred in 5.8% of the 874 patients receiving immediate-release sodium oxybate in adult clinical trials in another population.
Other psychiatric reactions reported in adult clinical trials in patients with narcolepsy administered LUMRYZ included irritability, emotional disorder, panic attack, agitation, delirium, and obsessive thoughts. Other neuropsychiatric reactions reported in adult clinical trials in patients with narcolepsy administered immediate-release sodium oxybate and in the postmarketing setting for immediate-release sodium oxybate include hallucinations, paranoia, psychosis, aggression, and agitation.
In a clinical trial in pediatric patients with narcolepsy administered immediate-release sodium oxybate, neuropsychiatric reactions, including acute psychosis, confusion, and anxiety were reported while taking immediate-release sodium oxybate.
The emergence or increase in the occurrence of behavioral or psychiatric events in patients taking LUMRYZ should be carefully monitored.
Parasomnias
Parasomnias can occur in patients taking LUMRYZ.
Sleepwalking, defined as confused behavior occurring at night and at times associated with wandering, was reported in 3% of 107 adult patients with narcolepsy treated with LUMRYZ. No patients treated with LUMRYZ discontinued due to sleepwalking. Sleepwalking was reported in 6% of 781 patients with narcolepsy treated with immediate-release sodium oxybate in adult controlled and long-term open-label studies, with <1% of patients discontinuing due to sleepwalking. In controlled trials, rates of sleepwalking were similar for patients taking placebo and patients taking immediate-release sodium oxybate. It is unclear if some or all of the reported sleepwalking episodes correspond to true somnambulism, which is a parasomnia occurring during non-REM sleep, or to any other specific medical disorder. Five instances of sleepwalking with potential injury or significant injury were reported during a clinical trial of immediate-release sodium oxybate in patients with narcolepsy.
Parasomnias, including sleepwalking, have been reported in a pediatric clinical trial and in postmarketing experience with immediate-release sodium oxybate. Therefore, episodes of sleepwalking should be fully evaluated, and appropriate interventions considered.
Use in Patients Sensitive to High Sodium Intake
LUMRYZ has a high sodium content. In patients sensitive to sodium intake (e.g., those with heart failure, hypertension, or renal impairment), consider the amount of daily sodium intake in each dose of LUMRYZ. Table 1 provides the approximate sodium content per LUMRYZ dose.
LUMRYZ Dose | Sodium Content/Total Nightly Exposure |
4.5 g per night | 820 mg |
6 g per night | 1100 mg |
7.5 g per night | 1400 mg |
9 g per night | 1640 mg |
6 ADVERSE REACTIONS
The following clinically significant adverse reactions appear in other sections of the labeling:
● CNS Depression [see Warnings and Precautions (5.1) ]
● Abuse and Misuse [see Warnings and Precautions (5.2) ]
● Respiratory Depression and Sleep-Disordered Breathing [see Warnings and Precautions (5.4) ]
● Depression and Suicidality [see Warnings and Precautions (5.5) ]
● Other Behavioral or Psychiatric Adverse Reactions [see Warnings and Precautions (5.6) ]
● Parasomnias [see Warnings and Precautions (5.7) ]
● Use in Patients Sensitive to High Sodium Intake [see Warnings and Precautions (5.8) ]
Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
Adult Patients
LUMRYZ was studied in one placebo-controlled trial (Study 1) [see Clinical Studies (14.1) ] in 212 patients with narcolepsy (107 patients treated with LUMRYZ and 105 with placebo).
Adverse Reactions Leading to Treatment Discontinuation
In Study 1, 15.9% of patients treated with LUMRYZ discontinued because of adverse reactions, compared to 1.9% of patients receiving placebo. The most common adverse reaction leading to discontinuation was dizziness (4.7%). For LUMRYZ, 5.6% of patients discontinued due to adverse reactions on 4.5 g, 4.1% on 6 g, 4.5% on 7.5 g, and 3.9% on 9 g dose.
Most Common Adverse Reactions
The most common adverse reactions (incidence ≥5% and greater than placebo) reported for any dose of LUMRYZ were nausea, dizziness, enuresis, headache, and vomiting.
Adverse Reactions Occurring at an Incidence of 2% or Greater
Table 2 lists adverse reactions occurring in 2% or more of LUMRYZ-treated patients on any individual dose and at a rate greater than placebo-treated patients in Study 1.
Adverse Reaction | Placebo (N=105) % | LUMRYZ 4.5 g (N=107) % | LUMRYZ 6 g (N=97) % | LUMRYZ 7.5 g (N=88) % | LUMRYZ 9 g (N=77) % |
Gastrointestinal Disorders | |||||
Vomiting | 2 | 3 | 3 | 6 | 5 |
Nausea | 3 | 6 | 8 | 7 | 1 |
Investigations | |||||
Weight Decreased | 0 | 1 | 0 | 0 | 4 |
Metabolism and Nutritional Disorders | |||||
Decreased Appetite | 0 | 4 | 4 | 3 | 3 |
Nervous System Disorders | |||||
Dizziness | 0 | 6 | 4 | 6 | 5 |
Somnolence | 1 | 0 | 1 | 2 | 4 |
Headache | 6 | 7 | 5 | 6 | 0 |
Psychiatric Disorders | |||||
Enuresis | 0 | 2 | 4 | 9 | 9 |
Anxiety | 1 | 3 | 1 | 3 | 1 |
Somnambulism | 0 | 1 | 2 | 0 | 0 |
Dose-Response Information
In the clinical trial in adult patients with narcolepsy, a dose-response relationship was observed for enuresis and somnolence.
Additional Adverse Reactions
Adverse reactions observed in clinical studies with immediate-release sodium oxybate (≥2%), but not observed in Study 1 at a frequency of higher than 2%, and which may be relevant for LUMRYZ: diarrhea, abdominal pain upper, dry mouth, pain, feeling drunk, peripheral edema, cataplexy, muscle spasms, pain in extremity, tremor, disturbance in attention, paresthesia, sleep paralysis, disorientation, irritability, and hyperhidrosis.
Pediatric Patients (7 Years of Age and Older)
The safety of LUMRYZ for the treatment of cataplexy or excessive daytime sleepiness in pediatric patients 7 years of age and older with narcolepsy is supported by an adequate and well-controlled trial of immediate-release sodium oxybate [see Clinical Studies (14.2) ] . Below is a display of adverse reactions of immediate-release sodium oxybate in this adequate and well-controlled trial.
In this trial, 104 patients aged 7 to 17 years (37 patients aged 7 to 11 years; 67 patients aged 12 to 17 years) with narcolepsy received immediate-release sodium oxybate for up to one year. This trial included an open-label safety continuation period in which eligible patients received immediate-release sodium oxybate for up to an additional 2 years. The median and maximum exposure across the entire trial were 371 and 987 days, respectively.
Adverse Reactions Leading to Treatment Discontinuation
In the pediatric clinical trial with immediate-release sodium oxybate, 7 of 104 patients reported adverse reactions that led to withdrawal from the study (hallucination, tactile; suicidal ideation; weight decreased; sleep apnea syndrome; affect lability; anger, anxiety, depression; and headache).
Adverse Reactions in the Pediatric Clinical Trial
The most common adverse reactions (≥5%) were nausea (20%), enuresis (19%), vomiting (18%), headache (17%), weight decreased (13%), decreased appetite (9%), dizziness (8%), and sleepwalking (6%).
Additional information regarding safety in pediatric patients appears in the following sections:
● Respiratory Depression and Sleep-Disordered Breathing [see Warnings and Precautions (5.4) ]
● Depression and Suicidality [see Warnings and Precautions (5.5) ]
● Other Behavioral or Psychiatric Adverse Reactions [see Warnings and Precautions (5.6) ]
● Parasomnias [see Warnings and Precautions (5.7) ]
The overall adverse reaction profile of immediate-release sodium oxybate in the pediatric clinical trial was similar to that seen in the adult clinical trial with immediate-release sodium oxybate. The safety profile in pediatric patients with LUMRYZ is expected to be similar to that of adult patients treated with LUMRYZ and to that of pediatric patients treated with immediate-release sodium oxybate.
Postmarketing Experience
The following adverse reactions have been identified during postapproval use of sodium oxybate. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure:
Arthralgia, decreased appetite, fall•, fluid retention, hangover, headache, hypersensitivity, hypertension, memory impairment, nocturia, panic attack, vision blurred, and weight decreased.
•The sudden onset of sleep in patients taking sodium oxybate, including in a standing position or while rising from bed, has led to falls complicated by injuries, in some cases requiring hospitalization.
7 DRUG INTERACTIONS
Alcohol, Sedative Hypnotics, and CNS Depressants
LUMRYZ is contraindicated for use in combination with alcohol or sedative hypnotics. Use of other CNS depressants may potentiate the CNS-depressant effects of LUMRYZ [see Warnings and Precautions (5.1) ] . In addition to coadministration of LUMRYZ and alcohol being contraindicated because of respiratory depression, consumption of alcohol while taking LUMRYZ may also result in a more rapid release of the dose of sodium oxybate [see Clinical Pharmacology (12.3) ] .
11 DESCRIPTION
Sodium oxybate, a CNS depressant, is the active ingredient in LUMRYZ for extended-release oral suspension. The chemical name for sodium oxybate is sodium 4-hydroxybutyrate. The molecular formula is C 4 H 7 NaO 3 , and the molecular weight is 126.09 g/mole. The chemical structure is:
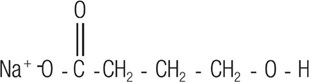
Sodium oxybate is a white to off-white solid powder.
Each packet of LUMRYZ contains 4.5 g, 6 g, 7.5 g, or 9 g of sodium oxybate, equivalent to 3.7 g, 5.0 g, 6.2 g, or 7.4 g of oxybate, respectively. The inactive ingredients are carrageenan, hydrogenated vegetable oil, hydroxyethyl cellulose, magnesium stearate, malic acid, methacrylic acid copolymer, microcrystalline cellulose, povidone, and xanthan gum.
12 CLINICAL PHARMACOLOGY
Mechanism of Action
LUMRYZ is a CNS depressant. The mechanism of action of LUMRYZ in the treatment of narcolepsy is unknown. Sodium oxybate is the sodium salt of gamma-hydroxybutyrate (GHB), an endogenous compound and metabolite of the neurotransmitter GABA. It is hypothesized that the therapeutic effects of LUMRYZ on cataplexy and excessive daytime sleepiness are mediated through GABA B actions at noradrenergic and dopaminergic neurons, as well as at thalamocortical neurons.
Pharmacokinetics
Following oral administration of LUMRYZ, the plasma levels of GHB increased dose-proportionally for C max and more than dose-proportionally for area under the plasma concentration-time curve (AUC inf ) (respectively, 2-fold and 2.3-fold increases as total daily dose is doubled from 4.5 g to 9 g).
No clinically significant difference in oxybate pharmacokinetics was observed between a single 6 g dose of LUMRYZ and two 3 g doses of immediate-release sodium oxybate administered 4 hours apart.
Absorption
Following oral administration of a single 6 g dose of LUMRYZ, the peak plasma concentration (C max ) was 66 mcg/mL and the time to peak plasma concentration (T max ) was 1.5 hours.
Effect of Food
Administration of LUMRYZ immediately after a high-fat meal resulted in a mean reduction in C max and AUC of GHB by 33% and 14%, respectively; average T max increased from 0.5 hours to 1.5 hours [ see Dosage and Administration (2.3) ].
Effect of Ethanol
An in vitro study showed alcohol-induced dose-dumping of sodium oxybate from extended-release oral suspension at 1 hour in the presence of 40% alcohol, and approximately 60% increase of drug release at 2 hours in the presence of 20% alcohol [see Contraindications (4) and Warnings and Precautions (5.1) ] .
Effect of Water Temperature
An in vitro dissolution study showed that LUMRYZ mixed with hot water (90°C) resulted in a dose-dumping phenomenon for the release of sodium oxybate, whereas warm water (50°C) did not significantly affect the drug release from the extended-release suspension [ see Dosage and Administration (2.3) ].
Effect of Water Flavoring
An in vitro dissolution study showed that LUMRYZ mixed with water prepared with calorie-free drink mix (e.g., Crystal Light Raspberry Lemonade Drink Mix) or flavored water enhancer (e.g., Mio Fruit Punch Concentrate Liquid Water Enhancer) did not impact the drug release from the extended-release suspension [see Dosage and Administration (2.3) ].
Distribution
GHB is a hydrophilic compound with an apparent volume of distribution averaging 190 mL/kg to 384 mL/kg. At GHB concentrations ranging from 3 mcg/mL to 300 mcg/mL, less than 1% is bound to plasma proteins.
Elimination
Metabolism
Animal studies indicate that metabolism is the major elimination pathway for GHB, producing carbon dioxide and water via the tricarboxylic acid (Krebs) cycle, and secondarily by β-oxidation. The primary pathway involves a cytosolic NADP + -linked enzyme, GHB dehydrogenase, which catalyzes the conversion of GHB to succinic semialdehyde, which is then biotransformed to succinic acid by the enzyme succinic semialdehyde dehydrogenase. Succinic acid enters the Krebs cycle where it is metabolized to carbon dioxide and water. A second mitochondrial oxidoreductase enzyme, a transhydrogenase, also catalyzes the conversion to succinic semialdehyde in the presence of α-ketoglutarate. An alternate pathway of biotransformation involves β-oxidation via 3,4-dihydroxybutyrate to carbon dioxide and water. No active metabolites have been identified.
Excretion
The clearance of GHB is almost entirely by biotransformation to carbon dioxide, which is then eliminated by expiration. On average, less than 5% of unchanged drug appears in human urine within 6 to 8 hours after dosing. Fecal excretion is negligible. GHB has an elimination half-life of 0.5 to 1 hour.
Specific Population
Geriatric Patients
There is limited experience with LUMRYZ in the elderly. Results from a pharmacokinetic study of immediate-release sodium oxybate (n=20) in another studied population indicate that the pharmacokinetic characteristics of GHB are consistent among younger (age 48 to 64 years) and older (age 65 to 75 years) adults.
Pediatric Patients
The pharmacokinetics of LUMRYZ has not been directly evaluated in pediatric patients.
The pharmacokinetics of immediate-release sodium oxybate were evaluated in pediatric patients 7 to 17 years of age (n=29). The pharmacokinetic characteristics of immediate-release sodium oxybate were shown to be similar in adults and pediatric patients. Body weight was found to be the major intrinsic factor affecting oxybate pharmacokinetics.
Male and Female Patients
In a study of 18 female and 18 male healthy adult volunteers, no gender differences were detected in the pharmacokinetics of GHB following an immediate-release 4.5 g oral dose of sodium oxybate.
Racial or Ethnic Groups
There are insufficient data to evaluate any pharmacokinetic differences among races.
Patients with Renal Impairment
No pharmacokinetic study in patients with renal impairment has been conducted.
Patients with Hepatic Impairment
The pharmacokinetics of GHB in 16 cirrhotic patients, half without ascites (Child’s Class A) and half with ascites (Child’s Class C), were compared to the kinetics in 8 subjects with normal hepatic function, after a single sodium oxybate oral dose of 25 mg/kg. AUC values were doubled in cirrhotic patients, with apparent oral clearance reduced from 9.1 mL/min/kg in healthy adults to 4.5 and 4.1 mL/min/kg in Class A and Class C patients, respectively. Elimination half-life was significantly longer in Class C and Class A patients than in control patients (mean t 1/2 of 59 minutes and 32 minutes, respectively, versus 22 minutes in control patients). LUMRYZ should not be initiated in patients with liver impairment [see Use in Specific Populations (8.6) ].
Drug Interaction Studies
In vitro studies with pooled human liver microsomes indicate that sodium oxybate does not significantly inhibit the activities of the human isoenzymes CYP1A2, CYP2C9, CYP2C19, CYP2D6, CYP2E1, or CYP3A, up to the concentration of 3 mM (378 mcg/mL), a level considerably higher than levels achieved with the maximum recommended dose.
A drug interaction study in healthy adults (age 18 to 55 years) was conducted with LUMRYZ and divalproex sodium. Co-administration of a single dose of LUMRYZ (6 g) with divalproex sodium ER at steady state resulted in an approximate 18% increase in AUC (90% CI ratio range of 112%-123%), which is not expected to be clinically meaningful, while C max was comparable. A single dose of LUMRYZ (6 g) did not appear to affect the pharmacokinetics of divalproex sodium. However, a pharmacodynamic interaction between LUMRYZ and divalproex sodium, a sedative antiepileptic drug, cannot be ruled out [see Warnings and Precautions (5.1) and Drug Interactions (7.1) ] .
Drug interaction studies in healthy adults (age 18 to 50 years) were conducted with immediate-release sodium oxybate and diclofenac and ibuprofen:
● Diclofenac: Co-administration of sodium oxybate (6 g per day as two equal doses of 3 grams dosed four hours apart) with diclofenac (50 mg/dose twice per day) showed no significant changes in systemic exposure to GHB. Co-administration did not appear to affect the pharmacokinetics of diclofenac.
● Ibuprofen: Co-administration of sodium oxybate (6 g per day as two equal doses of 3 grams dosed four hours apart) with ibuprofen (800 mg/dose four times per day also dosed four hours apart) resulted in comparable systemic exposure to GHB, as shown by plasma C max and AUC values. Co-administration did not affect the pharmacokinetics of ibuprofen.
Drug interaction studies in healthy adults demonstrated no pharmacokinetic interactions between immediate-release sodium oxybate and protriptyline hydrochloride, zolpidem tartrate, and modafinil. Also, there were no pharmacokinetic interactions with the alcohol dehydrogenase inhibitor fomepizole. However, pharmacodynamic interactions with these drugs cannot be ruled out. Alteration of gastric pH with omeprazole produced no significant change in the pharmacokinetics of GHB. In addition, drug interaction studies in healthy adults demonstrated no pharmacokinetic or clinically significant pharmacodynamic interactions between immediate-release sodium oxybate and duloxetine HCl.
13 NONCLINICAL TOXICOLOGY
Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Administration of sodium oxybate to rats at oral doses of up to 1,000 mg/kg/day for 83 (males) or 104 (females) weeks resulted in no increase in tumors. Plasma exposure (AUC) at the highest dose tested was 2 times that in humans at the maximum recommended human dose (MRHD) of 9 g per night.
The results of 2-year carcinogenicity studies in mouse and rat with gamma-butyrolactone, a compound that is metabolized to sodium oxybate in vivo, showed no clear evidence of carcinogenic activity. The plasma AUCs of sodium oxybate achieved at the highest doses tested in these studies were less than that in humans at the MRHD.
Mutagenesis
Sodium oxybate was negative in the in vitro bacterial gene mutation assay, an in vitro chromosomal aberration assay in mammalian cells, and in an in vivo rat micronucleus assay.
Impairment of Fertility
Oral administration of sodium oxybate (150, 350, or 1,000 mg/kg/day) to male and female rats prior to and throughout mating and continuing in females through early gestation resulted in no adverse effects on fertility. The highest dose tested is approximately equal to the MRHD on a mg/m 2 basis.
14 CLINICAL STUDIES
Cataplexy and Excessive Daytime Sleepiness (EDS) in Adult Narcolepsy
The effectiveness of LUMRYZ for the treatment of cataplexy or excessive daytime sleepiness (EDS) in adults with narcolepsy has been established based on a double-blind, randomized, placebo-controlled, two-arm multi-center study to assess the efficacy and safety of a once nightly administration of LUMRYZ in patients with narcolepsy (Study 1; NCT02720744 ).
A total of 212 patients were randomized to receive LUMRYZ or placebo in a 1:1 ratio and received at least one dose of study drug. The study was divided into four sequential study periods, and incorporated dose titration to stabilized dose administration of LUMRYZ (4.5 g, 6 g, 7.5 g, and 9 g). There was a three-week screening period, a 13-week treatment period including up-titration over a period of eight weeks, five weeks of stable dosing at 9 g/night, and a one-week follow-up period. Patients could be on concomitant stimulant as long as dosage was stable for 3 weeks prior to study start.
The three co-primary endpoints were the Maintenance of Wakefulness Test (MWT), Clinical Global Impression-Improvement (CGI-I), and mean change in weekly cataplexy attacks. The MWT measures latency to sleep onset (in minutes), averaged over five sessions at 2-hour intervals following nocturnal polysomnography. For each test session, patients were instructed to remain awake for as long as possible during 30-minute test sessions, and sleep latency was determined as the number of minutes patients could remain awake. The overall score was the mean sleep latency for the 5 sessions. The CGI-I was evaluated on a 7-point scale, centered at No Change, and ranging from Very Much Worse to Very Much Improved . Patients were rated by evaluators who based their assessments on the severity of narcolepsy at Baseline.
Demographic and mean baseline characteristics were similar for the LUMRYZ and placebo groups. A total of 76% were narcolepsy type 1 (NT1; with both symptoms of EDS and cataplexy) patients, and 24% were narcolepsy type 2 (NT2; with symptoms of EDS and without cataplexy) patients. The mean age was 31 years, and 68% were female. Approximately 63% of patients were on concomitant stimulant use. The mean MWT at baseline was 5 minutes for the LUMRYZ group, and 4.7 minutes for the placebo group. The mean number of cataplexy attacks per week at baseline was 18.9 in the LUMRYZ group and 19.8 in the placebo group. A statistically significant improvement was seen on the MWT, CGI-I, and mean weekly cataplexy attacks, for the 6 g (Week 3), 7.5 g (Week 8), and 9 g (Week 13) dose of LUMRYZ, compared to the placebo group (see Table 3, Table 4, and Table 5).
Dose | Treatment Group (N) | Change from Baseline (Minutes)• | Difference from Placebo [95% CI] | p-value |
•Mean MWT at baseline was 5.0 minutes for the LUMRYZ group and 4.7 minutes for the placebo group | ||||
6 g (Week 3) | LUMRYZ (87) | 8.1 | 5.0 [2.90;7.05] | <0.001 |
Placebo (88) | 3.1 | |||
7.5 g (Week 8) | LUMRYZ (76) | 9.6 | 6.2 [3.84;8.58] | <0.001 |
Placebo (78) | 3.3 | |||
9 g (Week 13) | LUMRYZ (68) | 10.8 | 6.1 [3.52;8.75] | <0.001 |
Placebo (78) | 4.7 | |||
Dose | Treatment Group (N) | Percentage of Responders (Much or Very Much Improved) | Odds Ratio [95% CI] | p-value |
6 g (Week 3) | LUMRYZ (87) | 40 | 10.3 [3.93;26.92] | <0.001 |
Placebo (87) | 6 | |||
7.5 g (Week 8) | LUMRYZ (75) | 64 | 5.7 [2.82;11.40] | <0.001 |
Placebo (81) | 22 | |||
9 g (Week 13) | LUMRYZ (69) | 73 | 5.6 [2.76;11.23] | <0.001 |
Placebo (79) | 32 |
Dose | Treatment Group (N) | Change from Baseline • | Difference from Placebo [95% CI] | p-value |
•Mean (SD) number of cataplexy attacks per week at baseline was 18.9 (8.7) in the LUMRYZ group and 19.8 (8.9) in the placebo group | ||||
6 g (Week 3) | LUMRYZ (73) | -7.4 | -4.8 [-7.03;-2.62] | <0.001 |
Placebo (72) | -2.6 | |||
7.5 g (Week 8) | LUMRYZ (66) | -10.0 | -6.3 [-8.74;-3.80] | <0.001 |
Placebo (69) | -3.7 | |||
9 g (Week 13) | LUMRYZ (55) | -11.5 | -6.7 [-9.32;-3.98] | <0.001 |
Placebo (62) | -4.9 | |||
Cataplexy and Excessive Daytime Sleepiness (EDS) in Pediatric Narcolepsy
The effectiveness of LUMRYZ in pediatric patients is based upon a clinical study in patients treated with immediate-release sodium oxybate, as described below.
The effectiveness of immediate-release sodium oxybate in the treatment of cataplexy or excessive daytime sleepiness in pediatric patients 7 years of age and older with narcolepsy was established in a double-blind, placebo-controlled, randomized-withdrawal study (NCT02221869 ). The study was conducted in 106 pediatric patients (median age: 12 years; range: 7 to 17 years) with a baseline history of at least 14 cataplexy attacks in a typical 2-week period prior to any treatment for narcolepsy symptoms. Of the 106 patients, 2 did not receive study drug and 63 patients were randomized 1:1 either to continued treatment with immediate-release sodium oxybate or to placebo. Randomization to placebo was stopped early as the efficacy criterion was met at the pre-planned interim analysis.
Patients entered the study either taking a stable dosage of immediate-release sodium oxybate or were immediate-release sodium oxybate-naïve. CNS stimulants were allowed at entry, and approximately 50% of patients continued taking a stable dose of stimulant throughout the stable-dose and double-blind periods. Immediate-release sodium oxybate-naïve patients were initiated and titrated based on body weight over a period of up to 10 weeks. The total nightly dose was administered in two divided doses, with the first dose given at nighttime and the second given 2.5 to 4 hours later. Once a stable dosage of immediate-release sodium oxybate had been achieved, these patients entered the 2-week stable-dose period; patients on a stable dosage of immediate-release sodium oxybate at study entry remained on this dosage for 3 weeks prior to randomization. Efficacy was established at dosages ranging from 3 g to 9 g of immediate-release sodium oxybate per night.
The primary efficacy measure was the change in frequency of cataplexy attacks. In addition, change in cataplexy severity was evaluated with the Clinical Global Impression of Change for cataplexy severity. The Clinical Global Impression of Change is evaluated on a 7-point scale, centered at No Change, and ranging from Very Much Worse to Very Much Improved. The efficacy of immediate-release sodium oxybate in the treatment of excessive daytime sleepiness in pediatric patients with narcolepsy was evaluated with the change in the Epworth Sleepiness Scale (Child and Adolescent) score. The Epworth Sleepiness Scale (Child and Adolescent) is a modified version of the Epworth Sleepiness Scale used in the adult clinical trial with immediate-release sodium oxybate. The Epworth Sleepiness Scale is intended to evaluate the extent of sleepiness in everyday situations by asking the patient a series of questions. In these questions, patients were asked to rate their chances of dozing during each of 8 activities on a scale from 0-3 (0=never; 1=slight; 2=moderate; 3=high). Higher total scores indicate a greater tendency to sleepiness. The overall change in narcolepsy condition was assessed by the Clinical Global Impression of Change for narcolepsy overall. Efficacy was assessed during or at the end of the 2-week double-blind treatment period, relative to the last 2 weeks or end of the stable-dose period (see Tables 6 and 7).
Pediatric patients taking stable dosages of immediate-release sodium oxybate who were withdrawn from immediate-release sodium oxybate treatment and were randomized to placebo during the double-blind treatment period experienced a statistically significant increase in weekly cataplexy attacks compared with patients who were randomized to continue treatment with immediate-release sodium oxybate. Patients randomized to receive placebo during the double-blind treatment period experienced a statistically significant worsening of EDS compared with patients randomized to continue receiving immediate-release sodium oxybate (see Table 6).
• For weekly number of cataplexy attacks, baseline value is calculated from the last 14 days of the stable-dose period. † For Epworth Sleepiness Scale score, baseline value is collected at the end of stable-dose period. ‡ Weekly number of cataplexy attacks is calculated from all days within the double-blind treatment period. § For Epworth Sleepiness Scale, value is collected at the end of the double-blind treatment period. ¶ P-value from rank-based analysis of covariance (ANCOVA) with treatment as a factor and rank baseline value as a covariate. •• One patient in each of the treatment groups did not have baseline ESS score available and were not included in this analysis. | ||||
Treatment Group | Baseline •,† | Double-blind Treatment Period ‡,§ | Median Change from Baseline | Comparison to Placebo (p-value ¶ ) |
Median Number of Cataplexy Attacks (attacks/week) | ||||
Placebo (n=32) | 4.7 | 21.3 | 12.7 | |
Immediate-release Sodium Oxybate (n=31) | 3.5 | 3.8 | 0.3 | <0.0001 |
Median Epworth Sleepiness Scale (Child and Adolescent) Score | ||||
Placebo (n=31 •• ) | 11 | 12 | 3 | |
Immediate-release Sodium Oxybate (n=30 •• ) | 8 | 9 | 0 | 0.0004 |
Patients randomized to receive placebo during the double-blind treatment period experienced a statistically significant worsening of cataplexy severity and narcolepsy overall according to the clinician’s assessment compared with patients randomized to continue receiving immediate-release sodium oxybate (see Table 7).
• Responses indicate change of severity or symptoms relative to receiving immediate-release sodium oxybate treatment at baseline. † Percentages based on total number of observed values. ‡ Two patients randomized to immediate-release sodium oxybate did not have the CGIc assessments completed and were excluded from the analysis. § P-value from Pearson’s chi-square test. | ||||
Worsened, % † | CGIc Cataplexy Severity • | CGIc Narcolepsy Overall • | ||
Placebo (n=32) | Immediate-release Sodium Oxybate (n=29) ‡ | Placebo (n=32) | Immediate-release Sodium Oxybate (n=29) ‡ | |
Much worse or very much worse | 66% | 17% | 59% | 10% |
p-value § | 0.0001 | <0.0001 | ||
16 HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
LUMRYZ is a blend of white to off-white granules for extended-release oral suspension in water. LUMRYZ is supplied in cartons and a 28-day starter pack.
Cartons : each carton contains either 7 or 30 packets of LUMRYZ, a mixing cup, Prescribing Information and Medication Guide, and Instructions for Use (see Table 8). Dose packets contain a single dose of LUMRYZ provided in 4.5 g, 6 g, 7.5 g, or 9 g doses.
Strength | Package Size | NDC Number |
4.5 g | 7 packets | NDC 13551-001-07 |
30 packets | NDC 13551-001-30 | |
6 g | 7 packets | NDC 13551-002-07 |
30 packets | NDC 13551-002-30 | |
7.5 g | 7 packets | NDC 13551-003-07 |
30 packets | NDC 13551-003-30 | |
9 g | 7 packets | NDC 13551-004-07 |
30 packets | NDC 13551-004-30 |
28-day Starter Pack : contains four 7-count cartons, each containing a mixing cup, Prescribing Information and Medication Guide, and Instructions for Use (see Table 9). Dose packets contain a single dose of LUMRYZ provided in 4.5 g, 6 g, or 7.5 g doses.
28-day Starter Pack | Strength | Package Size | NDC Number |
Week 1 | 4.5 g | 7 packets | NDC 13551-005-01 |
Week 2 | 6 g | 7 packets | |
Week 3 | 6 g | 7 packets | |
Week 4 | 7.5 g | 7 packets |
Storage
Keep out of reach of children.
LUMRYZ should be stored at 20°C to 25°C (68°F to 77°F); excursions permitted to 15°C to 30°C (59°F to 86°F) (see USP Controlled Room Temperature).
Suspensions should be consumed within 30 minutes.
Handling and Disposal
LUMRYZ is a Schedule III drug under the Controlled Substances Act. LUMRYZ should be handled according to state and federal regulations. It is safe to dispose of LUMRYZ down the sanitary sewer.
INSTRUCTIONS FOR USE

This Instructions for Use contains information on how to take LUMRYZ. Read this Instructions for Use before you (or your child) take LUMRYZ and each time you (or your child) get a refill. There may be new information.
This information does not take the place of talking to your doctor about your (or your child's) medical condition or your (or your child's) treatment. If you have questions, please talk with your doctor.

● Take (or give) 1 packet of LUMRYZ each day at bedtime.
● You will need to mix LUMRYZ with water before you take or give your child the dose.
● You (or your child) should avoid getting out of bed after taking LUMRYZ. Some people fall asleep within 5 minutes of taking LUMRYZ and most will fall asleep within 15 minutes. The time it takes to fall asleep might be different from night to night.
● Medicines that cause sleepiness should not be used while taking LUMRYZ.
● Do not use LUMRYZ with alcohol.
● Do not drive or operate heavy machinery within 6 hours of taking LUMRYZ. Those activities should not be done until you know how LUMRYZ affects you.
● Mix and take (or give) LUMRYZ within 30 minutes. If not taken or given within 30 minutes of mixing, throw it away (dispose of it) and prepare a new dose.

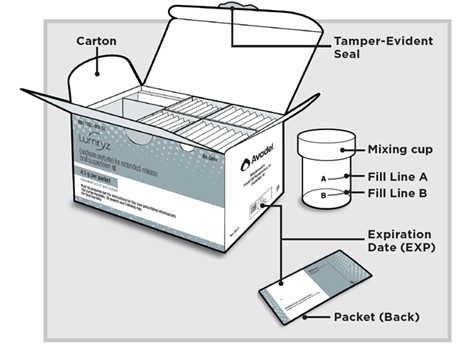
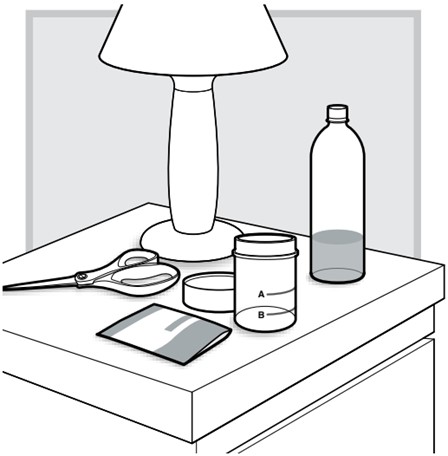
LUMRYZ comes in different package sizes. The LUMRYZ package you receive may look different from the picture shown above.
Additional supplies needed


● Store LUMRYZ at room temperature, between 68°F to 77°F (20°C to 25°C).
● Store LUMRYZ in the original packet prior to mixing with water.
● LUMRYZ suspension should be taken within 30 minutes of preparation.
● When you have finished using the LUMRYZ packet, throw it away (dispose of it) in the trash. If any LUMRYZ remains in the packet, rinse it down the sink before throwing away.
● Store LUMRYZ and all medicines out of the reach of children and pets.

● Before using a new LUMRYZ carton, check the tamper-evident seal on the carton lid to make sure it is not missing or broken.
● Do not use if the tamper-evident seal is missing or broken.
● Check the expiration date (EXP) on the LUMRYZ carton.
● Do not use LUMRYZ after the expiration date (EXP) on the label has passed.
● Open the LUMRYZ carton by tearing the tamper-evident seal with your hands or by using a pair of scissors.

● Clean the mixing cup by rinsing it with water and letting it dry before each use.
● Do not use a measuring device other than the mixing cup that comes in your LUMRYZ carton to measure and take a dose of LUMRYZ.
● Check the expiration date (EXP) on the packet label. Do not use the LUMRYZ packet after the expiration date (EXP) has passed.
Important: Make sure to prepare LUMRYZ at bedside.


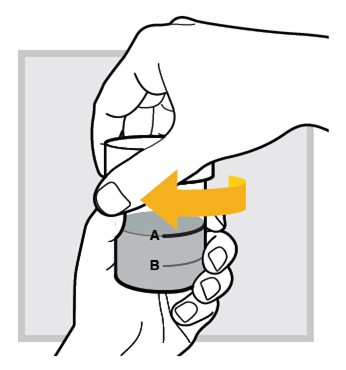
1.) At your (or your child's) bedside, open the mixing cup by twisting the cap to the left (counter-clockwise) to remove it.
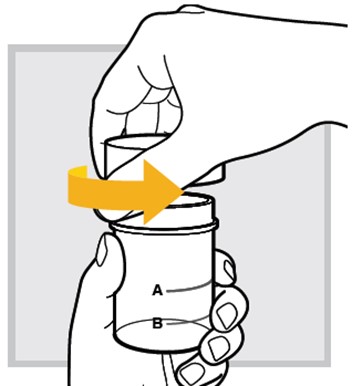
2.) Fill the mixing cup with water up to Fill Line A (top line) and set the mixing cup down on a flat surface.
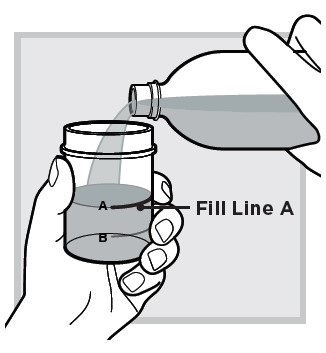
3.) Open 1 packet:
Use scissors to cut open the packet along the Cutting Line , located on the back of the packet.
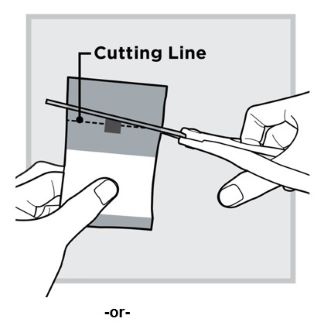
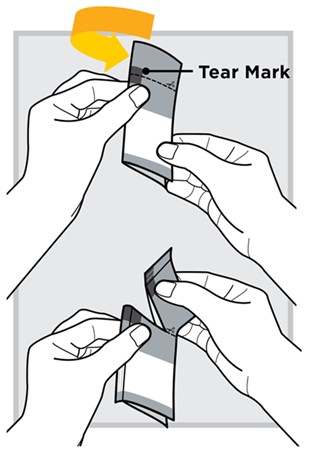
Fold the packet in half at the gray Tear Mark located on the back of the packet.
Tear the packet open with your hands.
4.) Pour the entire content from the packet into the water-filled mixing cup.
Make sure there is no powder left in the packet.

5.) Close the mixing cup by twisting the cap to the right (clockwise) until firmly closed.
6.) Mix the water and powder solution by shaking the closed mixing cup well for at least 60 seconds (1 minute) .
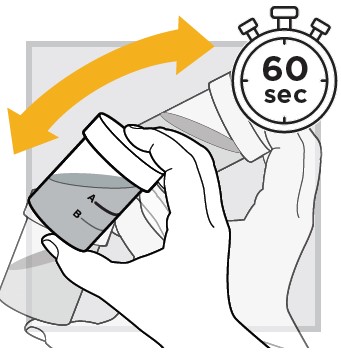
7.) Make sure the solution is mixed thoroughly.
The mixed solution will appear slightly milky with some lumps.
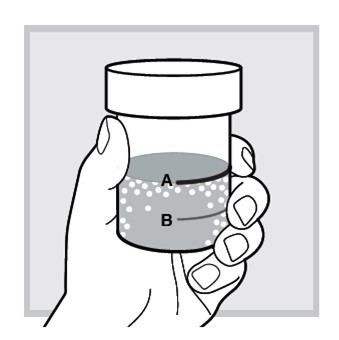
The mixing cup cap is not child resistant. If the mixed solution is not drank immediately, then do not remove the cap, and keep out of reach of children.

8.) Open the mixing cup by twisting the cap to the left (counter-clockwise) and remove it.
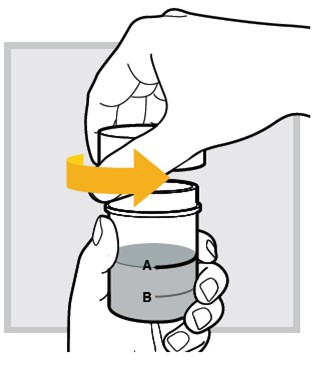
9.) While sitting in bed drink (or have your child drink) the mixed solution within 30 minutes of mixing.
Make sure you (or your child) drink all the mixed solution in the mixing cup.
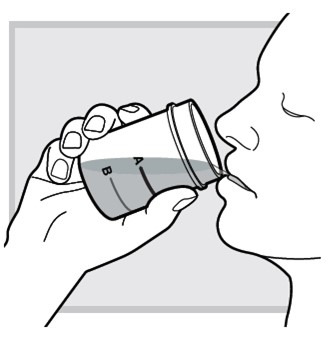
10.) Immediately refill the mixing cup with water up to Fill Line B (lower line) to mix in any medicine left in the mixing cup.
Do not open another packet of LUMRYZ. Take (or give) only 1 packet each day at bedtime.
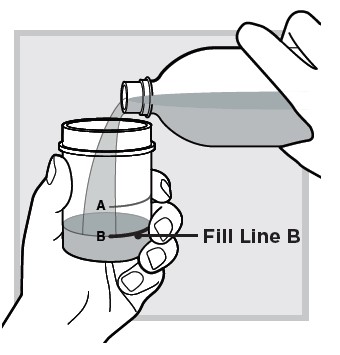
11.) Close the mixing cup by twisting the cap to the right (clockwise) until firmly closed.
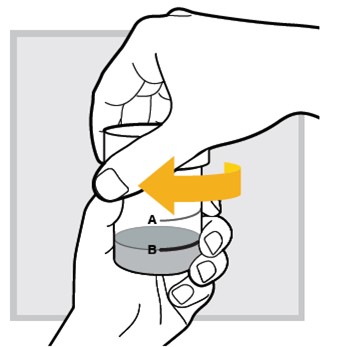
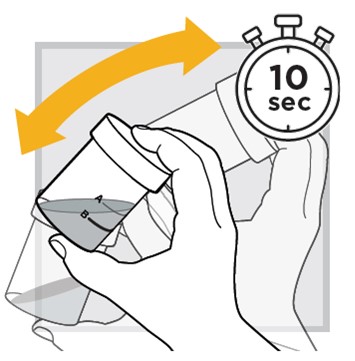
12.) Shake well again for 10 seconds .
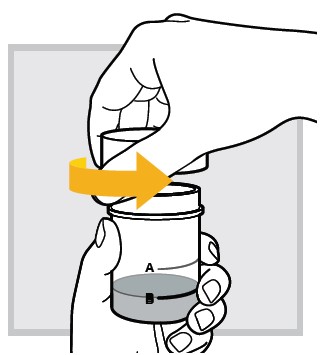
13.) Open the mixing cup by twisting the cap to the left (counter-clockwise) and remove it.
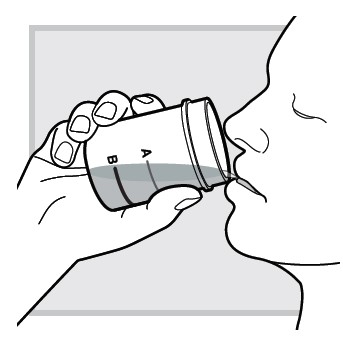
14.) Drink (or have your child drink) the mixed solution immediately after mixing.
Make sure you (or your child) drink all the mixed solution in the mixing cup.
15.) Leave the empty mixing cup at your (or your child's) bedside and immediately lie down (or have your child lie down) to go to sleep.
Avoid getting out of your bed (or having your child get out of bed) after taking LUMRYZ.


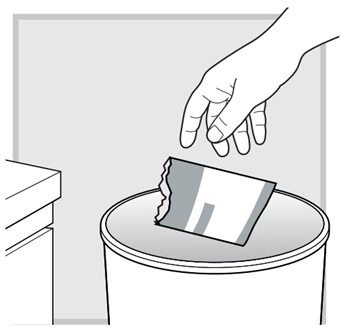
16.) The next day, place the empty LUMRYZ packet in the trash.
If any LUMRYZ remains in the packet, rinse it down the sink before (prior to) throwing away (disposal).
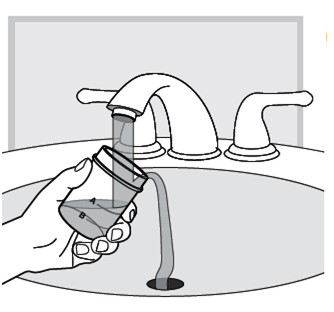
17.) Empty any unused LUMRYZ down the sink drain the next day.
Clean the mixing cup by rinsing it with water and letting it dry before each use.
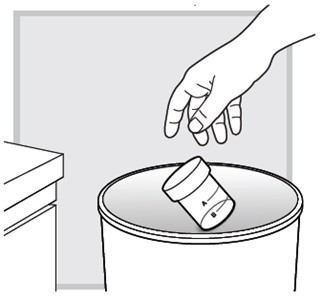
After you (or your child) finish all of the packets in the LUMRYZ carton
After you have (or your child has) finished your (or your child's) last packet in the carton, throw away the rinsed mixing cup in the trash.
If you have additional questions about LUMRYZ, talk with your doctor.
You can also contact:
Avadel CNS Pharmaceuticals, LLC
Chesterfield, MO 63005 USA
For more information on LUMRYZ,
visit www.lumryz.com or call
888-8AVADEL (888-828-2335).

© Avadel 2025. All rights reserved. AVADEL, the AVADEL logo, LUMRYZ, and the LUMRYZ logo are trademarks of an Avadel company.
This Instructions for Use has been approved by the U.S. Food and Drug Administration.
Revised: 11/2025