Get your patient on Truqap (Capivasertib)
Truqap patient education
Patient toolkit
Dosage & administration

Truqap prescribing information
INDICATIONS AND USAGE
TRUQAP is a kinase inhibitor indicated:
HR positive, HER2 negative locally advanced or metastatic breast cancer
- in combination with fulvestrant for the treatment of adult patients with hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative, locally advanced or metastatic breast cancer with one or more PIK3CA/AKT1/PTEN -alterations as detected by an FDA-authorized test following progression on at least one endocrine-based regimen in the metastatic setting or recurrence on or within 12 months of completing adjuvant therapy. (1.1 )
PTEN-deficient metastatic androgen pathway modulation-naïve or -sensitive (mAPMN/S) prostate cancer
- in combination with abiraterone and prednisone for the treatment of adult patients with metastatic androgen pathway modulation-naïve or -sensitive (mAPMN/S) prostate cancer that is PTEN-deficient as detected by an FDA-authorized test. (1.2 )
HR-positive, HER2-negative locally advanced or metastatic breast cancer
TRUQAP, in combination with fulvestrant, is indicated for the treatment of adult patients with hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative, locally advanced or metastatic breast cancer with one or more PIK3CA/AKT1/PTEN -alteration as detected by an FDA-authorized test following progression on at least one endocrine-based regimen in the metastatic setting or recurrence on or within 12 months of completing adjuvant therapy.
1.2 PTEN-deficient metastatic androgen pathway modulation-naïve or -sensitive prostate cancer
TRUQAP, in combination with abiraterone and prednisone, is indicated for treatment of adult patients with metastatic androgen pathway modulation-naïve or -sensitive (mAPMN/S) prostate cancer that is PTEN-deficient as detected by an FDA-authorized test.
DOSAGE AND ADMINISTRATION
- Select patients for the treatment of HR-positive, HER2-negative advanced or metastatic breast cancer with TRUQAP based on the presence of one or more of the following genetic alterations in tumor tissue: PIK3CA/AKT1/PTEN . (2.1 )
- Select patients for the treatment of mAPMN/S prostate cancer with TRUQAP based on PTEN deficiency in tumor tissue. (2.1 )
- Recommended Dosage: 400 mg orally twice daily, with or without food, for 4 days followed by 3 days off. (2.3 )
Patient Selection
Select patients for the treatment of HR-positive, HER2-negative advanced or metastatic breast cancer with TRUQAP, based on the presence of one or more of the following genetic alterations in tumor tissue: PIK3CA/AKT1/PTEN [see Clinical Studies (14) ] .
Select patients for the treatment of metastatic androgen pathway modulation-naïve or -sensitive (mAPMN/S) prostate cancer with TRUQAP based on PTEN deficiency in tumor tissue [see Clinical Studies (14) ] .
Information on FDA-authorized tests for patient selection for the detection of PIK3CA, AKT1, and PTEN alterations or PTEN deficiency are available at: http://www.fda.gov/CompanionDiagnostics
Recommended Evaluation Before Initiating TRUQAP
Evaluate fasting blood glucose (FG) and hemoglobin A1C (HbA1C) prior to starting TRUQAP and at regular intervals during treatment [see Warnings and Precautions (5.1) ] .
Recommended Dosage
The HR positive, HER2 negative locally advanced or metastatic breast cancer
The recommended dosage of TRUQAP, in combination with fulvestrant, is 400 mg orally twice daily (approximately 12 hours apart) with or without food, for 4 days followed by 3 days off. Continue TRUQAP until disease progression or unacceptable toxicity.
For premenopausal and perimenopausal women, administer a luteinizing hormone releasing hormone (LHRH) agonist according to current clinical practice standards.
For men, consider administering a LHRH agonist according to current clinical practice standards.
PTEN-deficient metastatic androgen pathway modulation-naïve or -sensitive (mAPMN/S) prostate cancer
The recommended dosage of TRUQAP, in combination with abiraterone and prednisone, is 400 mg orally twice daily (approximately 12 hours apart) with or without food, for 4 days followed by 3 days off. Continue TRUQAP until disease progression or unacceptable toxicity.
Patients with mAPMN/S prostate cancer should also receive a gonadotropin-releasing hormone (GnRH) analog concurrently or should have had bilateral orchiectomy.
Refer to the Prescribing Information of the individual therapeutic agents used in combination with TRUQAP for dosing and administration information.
Administration
TRUQAP dosing schedule for each week is provided in Table 1.
Day | 1 | 2 | 3 | 4 | 5 No dosing on day 5, 6 and 7 | 6 | 7 |
Morning | 2 x 200 mg | 2 x 200 mg | 2 x 200 mg | 2 x 200 mg | |||
Evening | 2 x 200 mg | 2 x 200 mg | 2 x 200 mg | 2 x 200 mg |
Swallow TRUQAP tablets whole. Do not chew, crush, or split tablets prior to swallowing. Do not take tablets that are broken, cracked, or otherwise not intact.
If a patient misses a dose within 4 hours of the scheduled time, instruct the patient to take the missed dose. If a patient misses a dose more than 4 hours of the scheduled time, instruct the patient to skip the dose and take the next dose at its usual scheduled time.
If a patient vomits a dose, instruct the patient not to take an additional dose and take the next dose at its usual scheduled time.
Dosage Modification for Adverse Reactions
The recommended dose reductions for adverse reactions are listed in Table 2. Permanently discontinue TRUQAP if unable to tolerate the second dose reduction.
| TRUQAP | Dose and Schedule |
|---|---|
First dose reduction | 320 mg twice daily for 4 days followed by 3 days off |
Second dose reduction | 200 mg twice daily for 4 days followed by 3 days off |
The recommended dosage modifications for adverse reactions are provided in Table 3.
Adverse Reaction | Severity Severity grading according to Common Terminology Criteria for Adverse Events (CTCAE) Version 5.0. | TRUQAP Dosage Modification |
Hyperglycemia For the management of suspected or confirmed diabetic ketoacidosis (DKA) see Warnings and Precautions (5.1) . (Fasting Glucose [FG]) | FG > ULN‑160 mg/dL or FG > ULN‑8.9 mmol/L or HbA1C > 7% | Consider initiation or intensification of oral anti-diabetic treatment. |
FG 161‑250 mg/dL or FG 9‑13.9 mmol/L | Withhold TRUQAP until FG decrease ≤ 160 mg/dL (or ≤ 8.9 mmol/L). If recovery occurs in ≤ 28 days, resume TRUQAP at same dose. If recovery occurs in > 28 days, resume TRUQAP at one lower dose. | |
FG 251‑500 mg/dL or FG 14‑27.8 mmol/L | Withhold TRUQAP until FG decrease ≤ 160 mg/dL (or ≤ 8.9 mmol/L). If recovery occurs in ≤ 28 days, resume TRUQAP at one lower dose. If recovery occurs in > 28 days, permanently discontinue TRUQAP. | |
FG > 500 mg/dL or FG > 27.8 mmol/L or life-threatening sequelae of hyperglycemia at any FG level | Withhold TRUQAP. For life-threatening sequelae of hyperglycemia or if FG persists at ≥ 500 mg/dL after 24 hours, permanently discontinue TRUQAP. If FG ≤ 500 mg/dL (or ≤ 27.8 mmol/L) within 24 hours, then follow the guidance in the table for the relevant grade. | |
Diarrhea | Grade 2 | Withhold TRUQAP until recovery to ≤ Grade 1. If recovery occurs in ≤ 28 days, resume TRUQAP at same dose or one lower dose as clinically indicated. If recovery occurs in > 28 days, resume at one lower dose as clinically indicated. For recurrence, reduce TRUQAP by one lower dose. |
Grade 3 | Withhold TRUQAP until recovery to ≤ Grade 1. If recovery occurs in ≤ 28 days, resume TRUQAP at same dose or one lower dose as clinically indicated. If recovery occurs in > 28 days, permanently discontinue TRUQAP. | |
Grade 4 | Permanently discontinue TRUQAP. | |
Cutaneous Adverse Reactions | Grade 2 | Withhold TRUQAP until recovery to ≤ Grade 1. Resume TRUQAP at the same dose. Persistent or recurrent: reduce TRUQAP by one lower dose. |
Grade 3 | Withhold TRUQAP until recovery to ≤ Grade 1. If recovery occurs in ≤ 28 days, resume TRUQAP at same dose. If recovery occurs in > 28 days, resume TRUQAP at one lower dose. For recurrent Grade 3, permanently discontinue TRUQAP. | |
Grade 4 | Permanently discontinue TRUQAP. | |
Other Adverse Reactions [see Adverse Reactions (6.1) ] | Grade 2 | Withhold TRUQAP until recovery to ≤ Grade 1. Resume TRUQAP at the same dose. |
Grade 3 | Withhold TRUQAP until recovery to ≤ Grade 1. If recovery occurs in ≤ 28 days, resume TRUQAP at same dose. If recovery occurs in > 28 days, resume TRUQAP at one lower dose. | |
Grade 4 | Permanently discontinue TRUQAP. |
Dosage Modifications for Strong and Moderate CYP3A Inhibitors
Avoid concomitant use with strong CYP3A inhibitors. If concomitant use with a strong CYP3A inhibitor cannot be avoided, reduce the dosage of TRUQAP to 320 mg orally twice daily for 4 days followed by 3 days off [see Drug Interactions (7.1) ] .
When concomitantly used with a moderate CYP3A inhibitor, reduce the dosage of TRUQAP to 320 mg orally twice daily for 4 days followed by 3 days off.
After discontinuation of a strong or moderate CYP3A inhibitor, resume the TRUQAP dosage (after 3 to 5 half-lives of the inhibitor) that was taken prior to initiating the strong or moderate CYP3A inhibitor.
DOSAGE FORMS AND STRENGTHS
Tablets:
- 160 mg: beige film-coated, round, biconvex tablets debossed with ‘CAV’ above ‘160’ on one side and plain on the reverse.
- 200 mg: beige film-coated, capsule-shaped, biconvex tablets debossed with ‘CAV 200’ on one side and plain on the reverse.
USE IN SPECIFIC POPULATIONS
Lactation: Advise not to breastfeed. (8.2 )
Pregnancy
Risk Summary
TRUQAP is used in combination with fulvestrant. Refer to the Full Prescribing Information of fulvestrant for pregnancy information.
Based on findings in animals and mechanism of action, TRUQAP can cause fetal harm when administered to a pregnant woman [see Clinical Pharmacology (12.1) ] . There are no available data on the use of TRUQAP in pregnant women. In an animal reproduction study, oral administration of capivasertib to pregnant rats during the period of organogenesis caused adverse developmental outcomes, including embryo-fetal mortality and reduced fetal weights at maternal exposures 0.7 times the human exposure (AUC) at the recommended dose of 400 mg twice daily ( see Data ). Advise pregnant women and females of reproductive potential of the potential risk to a fetus.
The background risk of major birth defects and miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects is 2% to 4% and of miscarriage is 15% to 20% of clinically recognized pregnancies in the U.S. general population.
Data
Animal Data
In an embryo-fetal development study, pregnant rats received oral doses of capivasertib up to 150 mg/kg/day during the period of organogenesis. Administration of capivasertib resulted in maternal toxicities (reduced body weight gain and food consumption, increased blood glucose) and adverse developmental outcomes, including embryo-fetal deaths (post-implantation loss), reduced fetal weights, and minor fetal visceral variations at a dose of 150 mg/kg/day (0.7 times the human exposure at the recommended dose of 400 mg twice daily based on AUC).
In a pre- and post-natal assessment, pregnant rats received oral doses of capivasertib up to 150 mg/kg/day from gestation day 6 through at least lactation day 6. Administration of 150 mg/kg/day resulted in reduced litter and pup weights.
Lactation
Risk Summary
TRUQAP is used in combination with fulvestrant. Refer to the Full Prescribing Information of fulvestrant for lactation information.
There are no data on the presence of capivasertib or its metabolites in human milk or their effects on milk production or the breastfed child. Capivasertib was detected in the plasma of suckling rat pups ( see Data ). Because of the potential for serious adverse reactions in a breastfed child, advise women not to breastfeed during treatment with TRUQAP.
Data
Animal Data
In a pre- and post-natal assessment, when capivasertib was administered to maternal rats during the lactation period, capivasertib was detected in plasma of suckling rat pups on lactation day 7 to 8 [see Use in Specific Populations (8.1) ] . Plasma concentrations in pups were up to 0.6% of concentrations in maternal plasma in the 150 mg/kg/day group.
Females and Males of Reproductive Potential
TRUQAP is used in combination with fulvestrant or abiraterone. Refer to the Full Prescribing Information of fulvestrant or abiraterone for contraception and infertility information.
TRUQAP can cause fetal harm when administered to pregnant women [see Use in Specific Populations (8.1) ] .
Pregnancy Testing
Verify pregnancy status of females of reproductive potential prior to initiating TRUQAP [see Use in Specific Populations (8.1) ] .
Contraception
Females
Advise females of reproductive potential to use effective contraception during treatment with TRUQAP and for 1 month after the last dose.
Males
Advise male patients with female partners of reproductive potential to use effective contraception during treatment with TRUQAP and for 4 months after the last dose.
Pediatric Use
The safety and effectiveness of TRUQAP have not been established in pediatric patients.
Geriatric Use
Of the 355 patients who received TRUQAP in CAPItello-291, 115 (32%) patients were ≥ 65 years of age and 24 (7%) patients were ≥ 75 years of age. No overall differences in the efficacy of TRUQAP were observed between patients ≥ 65 years of age and younger patients. Analysis of the safety of TRUQAP comparing patients ≥ 65 years of age to younger patients suggest a higher incidence of Grade 3 to 5 adverse reactions (57% versus 36%), dosage reductions (30% versus 15%), dose interruptions (57% versus 30%), and permanent discontinuations (23% versus 8%), respectively.
Of the 503 patients who received TRUQAP in CAPItello-281, 331 (66%) patients were ≥ 65 years of age and 85 (17%) patients were ≥ 75 years of age. No overall differences in the efficacy of TRUQAP were observed between patients ≥ 65 years of age and younger patients. Analysis of the safety of TRUQAP comparing patients ≥ 65 years of age to younger patients suggest a higher incidence of Grade 3 to 5 adverse reactions (77% versus 60%), dosage reductions (36% versus 24%), dose interruptions (72% versus 53%), and permanent discontinuations (27% versus 7%), respectively. The differences in Grade 3 or 4 adverse reactions between patients ≥ 65 years of age and younger patients was primarily driven by an increased incidence of hyperglycemia and hypokalemia.
Renal Impairment
No dosage modification is recommended for patients with mild to moderate (creatinine clearance (CLcr) 30 to 89 mL/min) renal impairment [see Clinical Pharmacology (12.3) ] .
TRUQAP has not been studied in patients with severe (CLcr 15 to 29 mL/min) renal impairment.
Hepatic Impairment
No dosage modification is recommended for patients with mild hepatic impairment (bilirubin ≤ upper limit of normal (ULN) and AST > ULN or bilirubin > 1 to 1.5x ULN and any AST) [see Clinical Pharmacology (12.3) ] .
Monitor patients with moderate (bilirubin > 1.5 to 3x ULN and any AST) hepatic impairment for adverse reactions due to potential increased capivasertib exposure [see Warnings and Precautions (5.1 , 5.2 , 5.3) ] .
TRUQAP has not been studied in patients with severe (bilirubin > 3x ULN and any AST) hepatic impairment.
CONTRAINDICATIONS
TRUQAP is contraindicated in patients with severe hypersensitivity to TRUQAP or any of its components.
WARNINGS AND PRECAUTIONS
- Hyperglycemia: TRUQAP can cause severe hyperglycemia, including diabetic ketoacidosis and fatal outcomes. Evaluate blood glucose levels and hemoglobin A1C prior to starting and at regular intervals during treatment. Withhold, reduce dose, or permanently discontinue TRUQAP based on severity. (2.2 , 2.5 , 5.1 )
- Diarrhea: TRUQAP caused diarrhea in most patients. Advise patients to increase oral fluids, start antidiarrheal treatment, and consult with a healthcare provider if diarrhea occurs while taking TRUQAP. Withhold, reduce dose, or permanently discontinue TRUQAP based on severity. (2.5 , 5.2 )
- Cutaneous Adverse Reactions: Monitor for signs and symptoms of cutaneous adverse reactions. Withhold, reduce dose, or permanently discontinue TRUQAP based on severity. (2.5 , 5.3 )
- Embryo-Fetal Toxicity: TRUQAP can cause fetal harm. Advise patients of potential risk to a fetus and to use effective contraception. Refer to the Full Prescribing Information of fulvestrant for pregnancy and contraception information. (5.4 , 8.1 , 8.3 )
Hyperglycemia
TRUQAP can cause severe hyperglycemia, including diabetic ketoacidosis and fatal outcomes. Withhold TRUQAP in clinical situations known to increase the risk of severe hyperglycemia or ketoacidosis (e.g., suspected serious infection or acute illness). Before initiating treatment with TRUQAP, test fasting glucose levels (FPG or FBG), HbA1C levels, and optimize fasting glucose.
After initiating treatment with TRUQAP, monitor or self-monitor fasting glucose levels on Day 3 or 4 of the dosing week during weeks 1, 2, 4, 6 and 8; then monthly while on treatment with TRUQAP; and as clinically indicated. Monitor HbA1C levels every 3 months during treatment with TRUQAP and as clinically indicated and manage changes as appropriate. Patients with a history of well-controlled Type 2 diabetes mellitus may require intensified anti-hyperglycemic treatment and require close monitoring of fasting glucose levels.
For patients who experience hyperglycemia during treatment with TRUQAP, monitor fasting glucose at least twice weekly, on days on and off TRUQAP, until fasting glucose decreases to baseline levels. During treatment with anti-diabetic medications, monitor fasting glucose at least once a week for 2 months, followed by once every 2 weeks, or as clinically indicated.
Consider consultation with a healthcare practitioner with expertise in the treatment of hyperglycemia and initiation of fasting glucose monitoring at home for patients who have risk factors for hyperglycemia or who experience hyperglycemia. Advise patients of the signs and symptoms of hyperglycemia and counsel patients on lifestyle changes.
Withhold TRUQAP immediately when ketoacidosis is suspected. If ketoacidosis is confirmed, permanently discontinue TRUQAP.
The safety of TRUQAP has not been established in patients with Type 1 diabetes or Type 2 diabetes that is uncontrolled or requiring insulin at baseline as these patients were excluded from clinical studies [see Clinical Studies (14.1 , 14.2 )] .
Based on the severity of the hyperglycemia, withhold, reduce dose, or permanently discontinue TRUQAP [see Dosage and Administration (2.5) ].
CAPItello 291
Increased fasting glucose from baseline occurred in 37% of patients treated with TRUQAP, including 11% of patients with Grade 2 (FG > 160 to 250 mg/dL), 2% with Grade 3 (FG > 250 to 500 mg/dL), and 1.1% with Grade 4 (FG > 500 mg/dL) events.
The median time to first occurrence of hyperglycemia was 15 days (range: 1 to 367). Dose reduction for hyperglycemia was required in 0.6% of patients and permanent discontinuation was required in 0.6% of patients.
Diabetic metabolic decompensation occurred in 0.6% of patients, including diabetic ketoacidosis in 0.3%.
In CAPItello-291, 12% (43/355) of patients who received TRUQAP had an anti-hyperglycemic medication regimen either initiated or changed during the study, including treatment with insulin in 4.8% (17/355) of patients.
CAPItello 281
Increased fasting glucose from baseline occurred in 69% of patients treated with TRUQAP, including 25% of patients with Grade 2 (FG > 160 to 250 mg/dL), 12% with Grade 3 (FG > 250 to 500 mg/dL), and 1.2% of patients with Grade 4 (FG > 500 mg/dL) events.
The median time to first occurrence of hyperglycemia was 71 days (range: 1 to 1454). Dose reduction for hyperglycemia was required in 8.7% of patients and permanent discontinuation was required in 2.4% of patients.
Diabetic ketoacidosis occurred in 1.2% of patients.
In CAPItello 281, 41% (204/503) of patients who received TRUQAP had an anti-hyperglycemic medication regimen either initiated or changed during the study, including treatment with insulin in 16% (81/503) of patients.
Diarrhea
TRUQAP can cause severe diarrhea associated with dehydration.
Monitor patients for signs and symptoms of diarrhea. Advise patients to increase oral fluids and start antidiarrheal treatment at the first sign of diarrhea while taking TRUQAP. Withhold, reduce dose, or permanently discontinue TRUQAP based on severity [see Dosage and Administration (2.5) ] .
CAPItello-291
Diarrhea occurred in 72% of patients. Grade 3 or 4 diarrhea occurred in 9% of patients. The median time to first occurrence was 8 days (range 1 to 519). In the 257 patients with diarrhea, 59% required anti-diarrheal medications to manage symptoms. Dose reductions were required in 8% of patients, and 2% of patients permanently discontinued TRUQAP due to diarrhea. In patients with Grade ≥ 2 diarrhea (n=93) with at least 1 grade improvement (n=89), median time to improvement from the first event was 4 days (range: 1 to 154).
CAPItello 281
In CAPItello 281, diarrhea of any grade occurred in 264 (52%) patients, Grade 3 occurred in 31 (6%) patients and Grade 4 occurred in 1 (0.2%) patient. The median time to first occurrence was 12 days (range: 3 to 48). In the study, dose reductions were required in 26 (5%) patients and 6 (1.2%) patients discontinued TRUQAP due to diarrhea. In the 264 patients with diarrhea, anti-diarrheal medication was required in 64% (170/264) of patients to manage diarrhea symptoms.
Cutaneous Adverse Reactions
TRUQAP can cause cutaneous adverse reactions, which can be severe, including erythema multiforme (EM), palmar-plantar erythrodysesthesia, and drug reaction with eosinophilia and systemic symptoms (DRESS).
Monitor patients for signs and symptoms of cutaneous adverse reactions. Early consultation with a dermatologist is recommended. Withhold, reduce dose, or permanently discontinue TRUQAP based on severity [see Dosage and Administration (2.5) ] .
CAPItello 291
Cutaneous adverse reactions occurred in 58% of patients. Grade 3 or 4 cutaneous adverse reactions occurred in 17% of patients receiving TRUQAP. EM occurred in 1.7% of patients, and DRESS occurred in 0.3% of patients. Dose reduction was required in 7% of patients and 7% of patients permanently discontinued TRUQAP due to cutaneous adverse reactions.
The median time to onset of cutaneous adverse reactions was 13 days (range 1 to 575 days). Among the 204 patients with cutaneous adverse reactions, 44% (90/204) required corticosteroid treatment. Of these, 37% (76/204) were treated with topical corticosteroids and 19% (39/204) with systemic corticosteroids. In patients with Grade ≥ 2 cutaneous adverse reaction (n=116) with at least 1 grade improvement (n=104), median time to improvement from the first event was 12 days (range 2 to 544).
CAPItello 281
Cutaneous adverse reactions occurred in 53% of patients. Grade 3 cutaneous adverse reactions occurred in 84 (17%) patients and Grade 4 occurred in 1 (0.2%) patient. In the study, dose reduction was required in 58 (12%) patients and 38 (8%) patients discontinued TRUQAP due to rash. The median time to onset of rash was 13 days (range: 11 to 63 days). Among the 265 patients with cutaneous adverse reactions, 80% (212/265) required treatment and 91% (242/265) recovered in the study. Of these, 54% (115/212) were treated with topical corticosteroids and 25% (54/212) with systemic corticosteroids.
Embryo-Fetal Toxicity
Based on findings from animals and mechanism of action, TRUQAP can cause fetal harm when administered to a pregnant woman [see Clinical Pharmacology (12.1) ] . In an animal reproduction study, oral administration of capivasertib to pregnant rats during the period of organogenesis caused adverse developmental outcomes, including embryo-fetal mortality, and reduced fetal weights at maternal exposures 0.7 times the human exposure (AUC) at the recommended dosage of 400 mg twice daily.
Advise pregnant women and females of reproductive potential of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with TRUQAP and for 1 month after the last dose. Advise male patients with female partners of reproductive potential to use effective contraception during treatment with TRUQAP and for 4 months after the last dose [see Use in Specific Populations (8.1 , 8.3) ] .
TRUQAP is used in combination with fulvestrant or abiraterone. Refer to the Full Prescribing Information of fulvestrant or abiraterone for pregnancy and contraception information.
ADVERSE REACTIONS
The following adverse reactions are also discussed in greater details in other sections of the labeling:
- Hyperglycemia [see Warnings and Precautions (5.1) ]
- Diarrhea [see Warnings and Precautions (5.2) ]
- Cutaneous Adverse Reactions [see Warnings and Precautions (5.3) ]
Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The safety population described in WARNINGS and PRECAUTIONS reflects exposure to TRUQAP 400 mg orally, twice a day for 4 days followed by 3 days off, in combination with fulvestrant, in 355 patients with metastatic breast cancer in CAPItello-291 until disease progression or unacceptable toxicity. Among the 355 patients who received TRUQAP in combination with fulvestrant, 52% were exposed for 6 months or longer, and 27% were exposed for greater than one year. In this safety population, the most common (≥ 20%) adverse reactions including laboratory abnormalities were diarrhea (72%), cutaneous adverse reactions (58%), increased random glucose (57%), decreased lymphocytes (47%), decreased hemoglobin (45%), increased fasting glucose (37%), nausea and fatigue (35% each), decreased leukocytes (32%), increased triglycerides (27%), decreased neutrophils (23%), increased creatinine (22%), vomiting (21%), and stomatitis (20%).
The safety population described in WARNINGS and PRECAUTIONS also reflects exposure to TRUQAP 400 mg orally, twice a day for 4 days followed by 3 days off, in combination with abiraterone, in 503 patients with mAPMN/S prostate cancer with PTEN deficiency in CAPItello-281 until disease progression or unacceptable toxicity.
CAPItello-291
The safety of TRUQAP was evaluated in CAPItello-291, a clinical trial including 288 adult patients (155 patients in TRUQAP with fulvestrant arm and 133 patients in placebo with fulvestrant arm) whose breast cancer had one or more PIK3CA/AKT1/PTEN -alterations [see Clinical Studies (14) ] . Among patients who received TRUQAP, 61% were exposed for 6 months or longer and 30% were exposed for greater than one year.
Of the 155 patients who received TRUQAP with fulvestrant, the median age was 58 years (range 36 to 84); female (99%); White (48%), Asian (31%), Black (1.3%), American Indian/Alaska Native (0.6%), and other races (19%).
Serious adverse reactions occurred in 18% of patients receiving TRUQAP with fulvestrant. The most common serious adverse reactions (≥ 1%) were cutaneous adverse reaction (3.9%), diarrhea and pneumonia (2.6% each), vomiting and pyrexia (1.9% each), hyperglycemia, hypersensitivity, fatigue, renal injury and second malignancy (1.3% each).
Fatal adverse reactions occurred in 1.3% of patients who received TRUQAP with fulvestrant, including sepsis (0.6%), and acute myocardial infarction (0.6%).
Permanent TRUQAP discontinuation due to an adverse reaction occurred in 10% of patients. The most common adverse reaction (≥ 2%) leading to permanent discontinuation of TRUQAP was cutaneous adverse reactions (6%). Dosage interruptions of TRUQAP due to an adverse reaction occurred in 39% of patients. Adverse reactions leading to dosage interruption in ≥ 2% of patients included cutaneous adverse reactions (14%), diarrhea (10%), pyrexia (4.5%), vomiting and nausea (3.2% each), and fatigue (2.6%).
Dose reductions of TRUQAP due to adverse reactions occurred in 21% of patients receiving TRUQAP with fulvestrant. Adverse reactions leading to TRUQAP dose reductions in ≥ 2% of patients were diarrhea and cutaneous adverse reactions (8% each).
The most common (≥ 20%) adverse reactions including laboratory abnormalities were diarrhea (77%), increased random glucose (58%), cutaneous adverse reaction (56%), decreased lymphocytes (49%), decreased hemoglobin (47%), fatigue (38%), increased fasting glucose (37%), nausea and decreased leukocytes (35% each), increased triglycerides (30%), stomatitis (25%), decreased neutrophils (25%), and vomiting (21%). Adverse reactions and laboratory abnormalities are listed in Table 4 and Table 5, respectively.
Adverse Reaction | TRUQAP with Fulvestrant N=155 | Placebo with Fulvestrant N=133 | ||
All Grades % | Grade 3 or 4 % | All Grades % | Grade 3 or 4 % | |
Gastrointestinal Disorders | ||||
Diarrhea | 77 | 12 | 19 | 0.8 |
Nausea | 35 | 1.3 | 14 | 0.8 |
Stomatitis Includes other related terms. | 25 | 1.9 | 5 | 0 |
Vomiting | 21 | 1.9 | 7 | 0.8 |
Skin and Subcutaneous Tissue Disorders | ||||
Cutaneous adverse reactions Cutaneous adverse reaction includes butterfly rash, dermatitis, allergic dermatitis, dry skin, eczema, erythema multiforme, hand dermatitis, palmar-plantar erythrodysesthesia syndrome, pruritus, rash, erythematous rash, maculo-papular rash, papular rash, skin discoloration, skin fissures, skin reaction, skin ulcer, urticaria, purpura, erythema and drug eruption. | 56 | 15 | 16 | 0.8 |
General Disorders and Administration Site Conditions | ||||
Fatigue | 38 | 1.9 | 27 | 1.5 |
Metabolism and Nutrition Disorders | ||||
Decreased appetite | 17 | 0 | 8 | 0.8 |
Nervous System Disorders | ||||
Headache | 17 | 0 | 13 | 0.8 |
Infections and Infestations | ||||
Urinary tract infection | 14 | 0.6 | 5 | 0 |
Renal and Urinary disorders | ||||
Renal injury Renal injury includes acute kidney injury, renal failure, renal impairment, glomerular filtration rate decreased, increased creatinine and proteinuria. | 11 | 2.6 | 1.5 | 0.8 |
Clinically relevant adverse reactions occurring in < 10% of patients treated with TRUQAP included anemia, pyrexia, dysgeusia, dyspepsia, pneumonia, decreased weight, hypersensitivity (including anaphylactic reaction).
Laboratory Abnormality | TRUQAP with Fulvestrant The denominator used to calculate the rate varied from 129 to 155 based on the number of patients with a baseline value and at least one post-treatment value. | Placebo with Fulvestrant The denominator used to calculate the rate varied from 109 to 131 based on the number of patients with a baseline value and at least one post-treatment value. | ||
|
| All Grades (%) | Grade 3 or 4 (%) | |
Glucose Metabolism | ||||
Increased random glucose | 58 | 9 | 17 | 0 |
Increased fasting glucose | 37 | 0.6 | 29 | 0 |
Hematology | ||||
Decreased lymphocytes | 49 | 11 | 14 | 2.3 |
Decreased hemoglobin | 47 | 2 | 22 | 2.3 |
Decreased leukocytes | 35 | 0.6 | 23 | 0 |
Decreased neutrophils | 25 | 1.9 | 16 | 0.8 |
Decreased platelets | 12 | 1.9 | 6 | 0.8 |
Other Categories | ||||
Increased triglycerides | 30 | 0.7 | 22 | 0.9 |
Increased alanine aminotransferase | 23 | 2.6 | 13 | 0 |
Electrolytes/Renal | ||||
Decreased corrected calcium | 19 | 0.6 | 8 | 0 |
Increased creatinine | 19 | 1.3 | 4.6 | 0.8 |
Decreased potassium | 17 | 4.5 | 8 | 0 |
CAPItello-281
The safety of TRUQAP was evaluated in CAPItello-281, a clinical trial including 1006 adult patients (503 patients in TRUQAP with abiraterone and prednisone/prednisolone arm and 503 patients in placebo with abiraterone and prednisone/prednisolone arm) with mAPMN/S prostate cancer with PTEN deficiency [see Clinical Studies (14) ] . Among the 503 patients who received TRUQAP, 74% were exposed for 6 months or longer, 58% were exposed for 12 months or longer, and 35% were exposed for more than 2 years.
Serious adverse reactions occurred in 47% of patients who received TRUQAP with abiraterone and prednisone. The most common serious adverse reactions (≥ 2%) were pneumonia (7%), cutaneous adverse reactions (5%), hyperglycemia (5%), hemorrhage (3.4%), urinary tract infection (3.2%), ischemic cerebro /cardiovascular events (3.2%), acute kidney injury (2.2%), and hypokalemia (2.2%).
Fatal adverse reactions occurred in 8% of patients who received TRUQAP with abiraterone and prednisone, including sepsis (1.2%), ischemic cerebro-/cardiovascular events (1%), hemorrhage (0.6%), pneumonia (0.4%), and diabetic ketoacidosis (0.2%).
Dosage interruptions of TRUQAP due to an adverse reaction occurred in 65% of patients. Adverse reactions leading to dosage interruption in ≥ 2% of patients included cutaneous adverse reactions (22%), hyperglycemia (15%), diarrhea (13%), pneumonia (4%), hypokalemia (3.4%), fatigue (3.2%), pyrexia (3.2%), vomiting (2.6%), urinary tract infection (2.4%), nausea (2%) and anemia (2%).
Permanent TRUQAP discontinuation due to an adverse reaction occurred in 20% of patients. The most common adverse reactions (≥ 2%) leading to treatment discontinuation were cutaneous adverse reactions (8%) and hyperglycemia (2.4%).
Dose reductions of TRUQAP due to adverse reactions occurred in 32% patients. Adverse reactions leading to TRUQAP dose reductions in ≥ 2% of patients were cutaneous adverse reactions (12%), hyperglycemia (9%), diarrhea (5%), and fatigue (2%).
The most common (≥ 20%) adverse reactions including laboratory abnormalities were increased fasting glucose, decreased hemoglobin, decreased lymphocytes, cutaneous adverse reactions, diarrhea, decreased potassium, increased creatinine, increased non-fasting glucose, increased alanine aminotransferase, increased triglycerides, increased aspartate aminotransferase, decreased sodium, and fatigue.
Adverse reactions and laboratory abnormalities are listed in Table 6 and Table 7, respectively.
Adverse Reaction | TRUQAP plus Abiraterone N=503 | Placebo plus Abiraterone N=503 | ||
All Grades % | Grade 3 or 4 % | All Grades % | Grade 3 or 4 % | |
Gastrointestinal Disorders | ||||
Diarrhea | 53 | 6 | 9 | 0.4 |
Nausea | 13 | 0.6 | 5 | 0.2 |
Skin and Subcutaneous Tissue Disorders | ||||
Cutaneous adverse reactions Cutaneous adverse reaction includes brachioradial pruritus, dermatitis, dermatitis acneiform, dermatitis allergic, dermatitis atopic, dermatitis exfoliative, dermatitis exfoliative generalized, drug eruption, dry skin, eczema, eczema asteatotic, eczema nummular, erythema, erythema ab igne, erythema multiforme, exfoliative rash, eye pruritus, eyelids pruritus, genital erythema, mucocutaneous rash, palmar-plantar erythrodysesthesia syndrome, pruritus, pruritus allergic, pruritus genital, purpura, rash, rash erythematous, rash macular, rash maculo-papular, rash papular, rash pruritic, rash pustular, skin exfoliation, skin fissures, skin ulcer, stoma site rash, urticaria, and urticaria papular. | 53 | 17 | 16 | 0.4 |
General Disorders and Administration Site Conditions | ||||
Fatigue Includes other related terms. | 26 | 1.2 | 19 | 1.2 |
Pyrexia | 11 | 1.2 | 3.2 | 0 |
Infections and Infestations | ||||
Urinary tract infection | 16 | 4.2 | 12 | 1.8 |
Pneumonia | 11 | 6 | 6 | 2.4 |
Metabolism and Nutrition Disorders | ||||
Decreased appetite | 10 | 1 | 4.2 | 0 |
Clinically relevant adverse reactions occurring in < 10% of patients treated with TRUQAP included vomiting, stomatitis, decreased weight, dyspepsia, dysgeusia, hypersensitivity, and diabetic ketoacidosis.
Laboratory Abnormality | TRUQAP plus Abiraterone The denominator used to calculate the rate varied from 472 to 500 based on the number of patients with a baseline value and at least one post-treatment value | Placebo plus Abiraterone The denominator used to calculate the rate varied from 485 to 500 based on the number of patients with a baseline value and at least one post-treatment value. | ||
|
| All Grades (%) | Grade 3 or 4 (%) | |
Chemistry | ||||
Increased fasting glucose | 70 | 14 | 50 | 3 |
Decreased potassium | 51 | 19 | 36 | 10 |
Increased creatinine | 49 | 7 | 24 | 3 |
Increased non-fasting glucose | 49 | 23 | 31 | 2.3 |
Increased triglycerides | 37 | 3 | 32 | 2.6 |
Increased alanine aminotransferase | 38 | 9 | 36 | 6 |
Increased aspartate aminotransferase | 36 | 7 | 37 | 4 |
Decreased sodium | 30 | 5 | 23 | 3.6 |
Hematology | ||||
Decreased hemoglobin | 61 | 8 | 46 | 3.2 |
Decreased lymphocytes | 58 | 17 | 36 | 9 |
DRUG INTERACTIONS
Effects of Other Drugs on TRUQAP
Table 8 describes drug interactions where concomitant use of another drug affects TRUQAP.
Strong CYP3A Inhibitors | |
Clinical Impact |
|
Prevention or Management |
|
Moderate CYP3A Inhibitors | |
Clinical Impact |
|
Prevention or Management |
|
Strong and Moderate CYP3A Inducers | |
Clinical Impact |
|
Prevention or Management |
|
DESCRIPTION
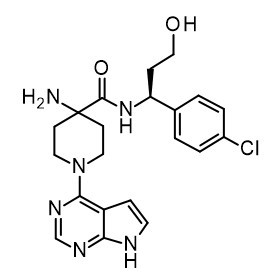
TRUQAP (capivasertib) is a kinase inhibitor. The molecular formula for capivasertib is C 21 H 25 ClN 6 O 2 and the molecular weight is 428.92 g/mol. The chemical name of capivasertib is 4-amino- N -[(1S)-1-(4-chlorophenyl)-3-hydroxypropyl]-1-(7 H -pyrrolo[2,3- d ]pyrimidin-4-yl)-4-piperidinecarboxamide. Capivasertib is freely soluble in water at pH values below 1.2 and practically insoluble at pH values above 6.8. Capivasertib has the following structural formula:
TRUQAP film-coated tablets are supplied for oral administration with 160 mg or 200 mg capivasertib. The tablets also contain croscarmellose sodium, dibasic calcium phosphate, magnesium stearate, and microcrystalline cellulose. The film coat contains the following inactive ingredients: copovidone, hypromellose, iron oxide black, iron oxide red, iron oxide yellow, medium chain triglycerides, polydextrose, polyethylene glycol 3350, and titanium dioxide.
CLINICAL PHARMACOLOGY
Mechanism of Action
Capivasertib is an inhibitor of all 3 isoforms of serine/threonine kinase AKT (AKT1, AKT2 and AKT3) and inhibits phosphorylation of downstream AKT substrates. AKT activation in tumors is a result of activation of upstream signaling pathways, mutations in AKT1 , loss of phosphatase and tensin homolog (PTEN) function and mutations in the catalytic subunit alpha of phosphatidylinositol 3-kinase ( PIK3CA ).
In vitro , capivasertib reduced growth of breast cancer cell lines including those with relevant PIK3CA or AKT1 mutations or PTEN alteration. In vivo , capivasertib alone and in combination with fulvestrant inhibited tumor growth of mouse xenograft models including estrogen receptor positive breast cancer models with alterations in PIK3CA, AKT1 , and PTEN .
In vitro , capivasertib reduced growth of AR-positive PTEN deficient prostate cancer cell lines. In vivo , capivasertib inhibited tumor growth in mouse xenograft models bearing PTEN-deficient prostate cancer cells and demonstrated increased antitumor activity when used in combination with abiraterone.
Pharmacodynamics
Exposure-Response Relationships
The exposure-response relationship and time course of pharmacodynamic response for the effectiveness of capivasertib have not been fully characterized. An exposure-response relationship was observed where increased capivasertib exposure was associated with increased incidence of diarrhea (CTCAE Grades 3 or 4) at doses of 80 to 800 mg (0.2 to 2 times the approved recommended dosage).
Cardiac Electrophysiology
At the recommended TRUQAP dose, a mean increase in the QTc interval > 20 ms was not observed.
Pharmacokinetics
Capivasertib pharmacokinetic parameters are presented as the geometric mean [geometric % coefficient of variation (%CV)], unless otherwise specified. The capivasertib steady-state AUC is 7,356 h·ng/mL (38%) and C max is 1,247 ng/mL (28%). Steady-state concentrations are predicted to be attained on the 3rd and 4th dosing day of each week, starting week 2.
Capivasertib plasma concentrations are approximately 0.2% to 5.1% of the steady state Cmax during the off-dosing days.
Capivasertib AUC and C max are proportional with dose over a range of 80 to 800 mg (0.2 to 2 times the approved recommended dosage).
Absorption
T max is approximately 1-2 hours. The absolute bioavailability is 29%.
Effect of Food
No clinically meaningful differences in capivasertib pharmacokinetics were observed following administration of TRUQAP with a high-fat meal (approximately 1,000 kcal; fat 60%) or a low-fat meal (approximately 400 kcal; fat 26%).
Distribution
The steady state oral volume of distribution is 1,847 L (36%). Capivasertib plasma protein binding is 78% and the plasma-to-blood ratio is 0.71.
Elimination
The half-life is 8.3 hours, and the steady-state oral clearance is 50 L/h (37% CV). Renal clearance was 21% of total clearance.
Metabolism
Capivasertib is primarily metabolized by CYP3A4 and UGT2B7.
Excretion
Following a single radiolabeled oral dose of 400 mg, the mean total recovery was 45% from urine and 50% from feces.
Specific Populations
No clinically significant differences in capivasertib pharmacokinetics were observed based on race/ethnicity (including White, Asian, Black, American Indian or Alaskan Native, and Native Hawaiian or Other Pacific Islander), sex (88% females), body weight (32 to 150 kg), age (26 to 87 years), mild hepatic impairment (bilirubin ≤ ULN and AST > ULN or bilirubin > 1 to 1.5x ULN), or mild to moderate renal impairment (CLcr 30 to 89 mL/min).
The effect of moderate (bilirubin > 1.5 to 3x ULN and any AST) hepatic impairment is not fully characterized.
TRUQAP has not been studied in patients with severe (bilirubin > 3x ULN and any AST) hepatic impairment or severe renal impairment (CLcr 15 to 29 mL/min).
Drug Interaction Studies
Clinical Studies and Model-Informed Approaches
Effect of Strong and Moderate CYP3A Inhibitors on Capivasertib: Itraconazole (strong CYP3A4 inhibitor) is predicted to increase capivasertib AUC by up to 1.7-fold and C max by up to 1.4-fold.
Erythromycin and verapamil (moderate CYP3A inhibitors) are predicted to increase capivasertib AUC by up to 1.5-fold and C max by up to 1.3-fold.
Effect of Strong and Moderate CYP3A Inducers on Capivasertib: Rifampicin (strong CYP3A4 inducer) is predicted to decrease capivasertib AUC by 70% and C max by 60%.
Efavirenz (moderate CYP3A4 inducer) is predicted to decrease capivasertib AUC by 60% and C max by 50%.
Effect of UGT2B7 Inhibitors on Capivasertib: Probenecid (UGT2B7 inhibitor) is not predicted to have a clinically meaningful effect on capivasertib pharmacokinetics.
Effect of Acid Reducing Agents on Capivasertib: Rabeprazole (gastric acid reducing agent) did not have a clinically meaningful effect on capivasertib pharmacokinetics.
Effect of Capivasertib on CYP3A Substrates: Concomitant use of TRUQAP increased midazolam (CYP3A substrate) AUC by 1.8-fold on day 4 and by 1.2-fold on day 7.
Effect of Capivasertib on CYP2D6 Substrates: TRUQAP is predicted to increase desipramine (CYP2D6 substrate) AUC by up to 2.1-fold on day 4.
Effect of Capivasertib on CYP2C9 Substrates: Concomitant use of TRUQAP with warfarin (CYP2C9 substrate) is not predicted to have a clinically meaningful effect on warfarin pharmacokinetics.
Effect of Capivasertib on UGT1A1 Substrates: TRUQAP is predicted to increase raltegravir (UGT1A1 substrate) AUC by up to 1.7-fold on day 4.
In-Vitro Studies
CYP450 Enzymes: Capivasertib induces CYP1A2.
Transporter Systems: Capivasertib inhibits BCRP, OATP1B1, OATP1B3, OAT3, MATE1, MATE2-K, and OCT2.
NONCLINICAL TOXICOLOGY
Carcinogenesis, Mutagenesis, Impairment of Fertility
In a 2-year rat carcinogenicity study, rats received oral doses up to 30 mg/kg/day in males and 75 mg/kg/day in females (approximately 0.2 times and 0.4 times the human exposure at the recommended dose based on AUC, respectively). While there were no statistically significant increases in tumors, there was an increase in malignant mesothelioma in the testis of male rats at 30 mg/kg/day, which is considered a rare tumor in the tested rat strain.
Capivasertib was genotoxic in the in vivo rat bone marrow micronucleus assay through an aneugenic mechanism. Capivasertib was not mutagenic in vitro in a bacterial reverse mutation (Ames) assay or mouse lymphoma gene mutation assay.
In repeat-dose toxicity studies up to 26 weeks duration in rats and 39 weeks duration in dogs, tubular degeneration in the testes and cellular debris in the epididymides were observed at oral capivasertib doses of 100 mg/kg/day in rats and 15 mg/kg/day in dogs (approximately 1 time the human exposure at the recommended dose of 400 mg twice daily based on AUC). In a male fertility study, capivasertib had no effect on fertility in male rats at oral doses up to 100 mg/kg/day following 10 weeks of treatment. Effects of capivasertib on female fertility have not been studied in animals.
CLINICAL STUDIES
Metastatic HR-Positive, HER2-Negative Breast Cancer
The efficacy of TRUQAP with fulvestrant was evaluated in CAPItello-291 (NCT04305496), a randomized, double-blind, placebo-controlled, multicenter trial that enrolled 708 adult patients with locally advanced (inoperable) or metastatic HR-positive, HER2-negative (defined as IHC 0 or 1+, or IHC 2+/ISH-) breast cancer of which 289 patients had tumors with eligible PIK3CA/AKT1/PTEN -alterations. Eligible PIK3CA/AKT1 activating mutations or PTEN loss of function alterations were identified in the majority of FFPE tumor specimens using FoundationOne®CDx next-generation sequencing (n=686). All patients were required to have progression on an aromatase inhibitor (AI) based treatment in the metastatic setting or recurrence on or within 12 months of completing (neo)adjuvant treatment with an AI. Patients could have received up to two prior lines of endocrine therapy and up to 1 line of chemotherapy for locally advanced (inoperable) or metastatic disease. Patients were excluded if they had clinically significant abnormalities of glucose metabolism (defined as patients with diabetes mellitus Type 1, Type 2 requiring insulin treatment, or HbA1c ≥8%).
Patients were randomized (1:1) to receive either 400 mg of TRUQAP (n=355) or placebo (n=353), given orally twice daily for 4 days followed by 3 days off treatment each week of 28-day treatment cycle. Fulvestrant 500 mg intramuscular injection was administered on cycle 1 days 1 and 15, and then at day 1 of each subsequent 28-day cycle. Patients were treated until disease progression, or unacceptable toxicity. Randomization was stratified by presence of liver metastases (yes vs. no), prior treatment with CDK4/6 inhibitors (yes vs. no) and geographical region (region 1: US, Canada, Western Europe, Australia, and Israel vs region 2: Latin America, Eastern Europe and Russia vs Region 3: Asia).
The major efficacy outcomes were investigator-assessed progression-free survival (PFS) in the overall population, and in the population of patients whose tumors have PIK3CA/AKT1/PTEN -alterations evaluated according to Response Evaluation Criteria in Solid Tumors (RECIST), version 1.1. Additional efficacy outcome measures were overall survival (OS), investigator assessed objective response rate (ORR) and duration of response (DoR).
Of the 289 patients whose tumors were PIK3CA/AKT1/PTEN -altered, the median age was 59 years (range 34 to 90); female (99%); White (52%), Asian (29%), Black (1%), American Indian/Alaska Native (0.7%), other races (17%) and 9% were Hispanic/Latino. Eastern Cooperative Oncology Group (ECOG) performance status was 0 (66%) or 1 (34%), and 18% were premenopausal or perimenopausal. Seventy-six percent of patients had an alteration in PIK3CA , 13% had an alteration in AKT1 , and 17% had an alteration in PTEN . All patients received prior endocrine-based therapy (100% AI based treatment and 44% received tamoxifen). Seventy-one percent of patients were previously treated with a CDK4/6 inhibitor and 18% received prior chemotherapy for locally advanced (inoperable) or metastatic disease.
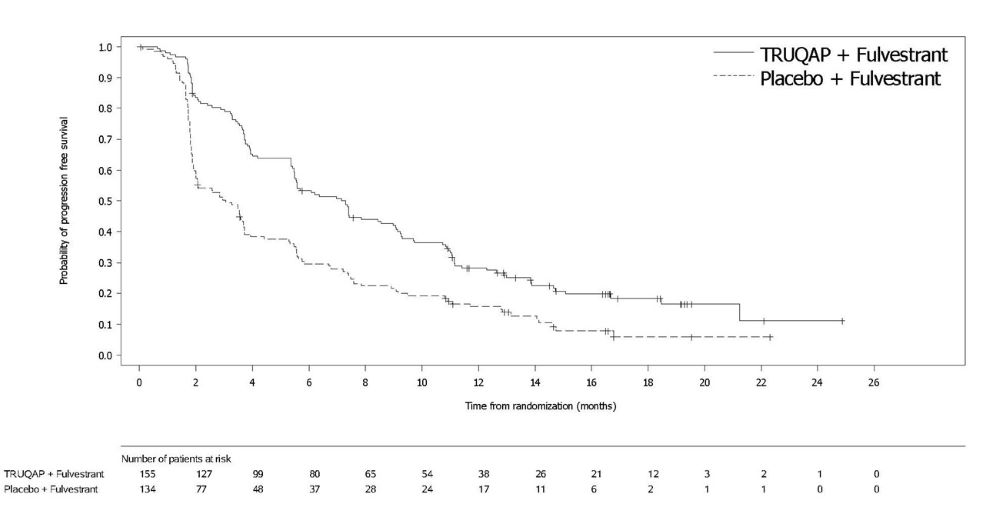
A statistically significant difference in PFS was observed in the overall population and the population of patients whose tumors have PIK3CA/AKT1/PTEN -alteration. An exploratory analysis of PFS in the 313 (44%) patients whose tumors did not have a PIK3CA/AKT1/PTEN -alteration showed a HR of 0.79 (95% CI: 0.61, 1.02), indicating that the difference in the overall population was primarily attributed to the results seen in the population of patients whose tumors have PIK3CA/AKT1/PTEN -alteration.
Efficacy results for PIK3CA/AKT1/PTEN -altered subgroup are presented in Table 9 and Figure 1. Results from the blinded independent review committee (BICR) assessment were consistent with the investigator assessed PFS results. Overall survival results were immature at the time of the PFS analysis (30% of the patients died).
| TRUQAP with fulvestrant N=155 | Placebo with fulvestrant N=134 | |
|---|---|---|
Investigator-Assessed Progression-Free Survival (PFS) | ||
| 121 (78%) | 115 (86%) |
| 7.3 (5.5, 9.0) | 3.1 (2.0, 3.7) |
| 0.50 (0.38, 0.65) | |
| <0.0001 | |
Investigator-Assessed Confirmed Objective Response Rate (ORR) | ||
| 132 | 124 |
| 26% (19, 34) | 8% (4, 14) |
| 2.3% | 0 |
| 23% | 8% |
| 10.2 (7.7, NC NC = not calculable ) | 8.6 (3.8, 9.2) |
Figure 1: Kaplan-Meier Plot of Progression-Free Survival in CAPItello-291 (Investigator Assessment, Patients with PIK3CA/AKT1/PTEN -Altered Tumors)
PTEN-Deficient Metastatic Androgen Pathway Modulation-Naïve or -Sensitive (mAPMN/S) Prostate Cancer
The efficacy of TRUQAP with abiraterone was evaluated in CAPItello-281 (NCT04305496), a randomized, double-blind, placebo-controlled, multicenter trial that enrolled 1012 adult patients with newly diagnosed, PTEN-deficient mAPMN/S prostate cancer (previously referred to as metastatic hormone-sensitive prostate cancer or mHSPC). Tumor PTEN deficiency status (≥ 90% of viable malignant cells with no specific cytoplasmic staining) was prospectively determined using central testing with the immunohistochemistry based VENTANA PTEN (SP218) RxDx Assay. The only allowable prior systemic therapy was up to 93 days of androgen deprivation therapy (ADT) and/or abiraterone. Patients were excluded if they had clinically significant abnormalities of glucose metabolism (defined as patients with diabetes mellitus Type 1 or Type 2 requiring insulin treatment, or HbA1c ≥ 8%.
Patients were randomized (1:1) to receive capivasertib 400 mg orally twice daily for 4 days followed by 3 days off treatment each week, in combination with abiraterone 1000 mg orally daily (N=507) compared with placebo plus abiraterone (N=505) until disease progression, death, withdrawal of consent, or unacceptable toxicity. Patients were required to receive either prednisone or prednisolone 5 mg daily, and a gonadotropin-releasing hormone (GnRH) analog or prior orchiectomy. Randomization was stratified by combination of volume of disease and visceral metastases (high-volume disease with visceral metastases vs. high-volume disease without visceral metastases vs. low-volume disease), and geographical region (Western Europe, Australia, North America, Latin America, Eastern Europe, Asia).
The major efficacy outcome was investigator-assessed radiographic progression-free survival (rPFS). Overall survival (OS) was an additional efficacy outcome measure.
Of the 1012 patients enrolled, the median age was 68 years (range 42 to 88); 52% were White, 37% Asian, 4% American Indian or Alaska Native, 4% Not reported, 1% Black, 1% Missing and 0.4% Other; 13% were Hispanic or Latino, 86% Not Hispanic or Latino, 1% Missing; and baseline Eastern Cooperative Oncology Group (ECOG) performance status was 0 (64%), 1 (36%). Seventy-four percent of patients had high volume disease including 19% with visceral metastases and 55% without visceral metastases. Type 2 diabetes mellitus was reported in 14% of patients. Ninety-nine percent of patients received previous hormonal therapy including 98% with ADT and 11% with abiraterone.
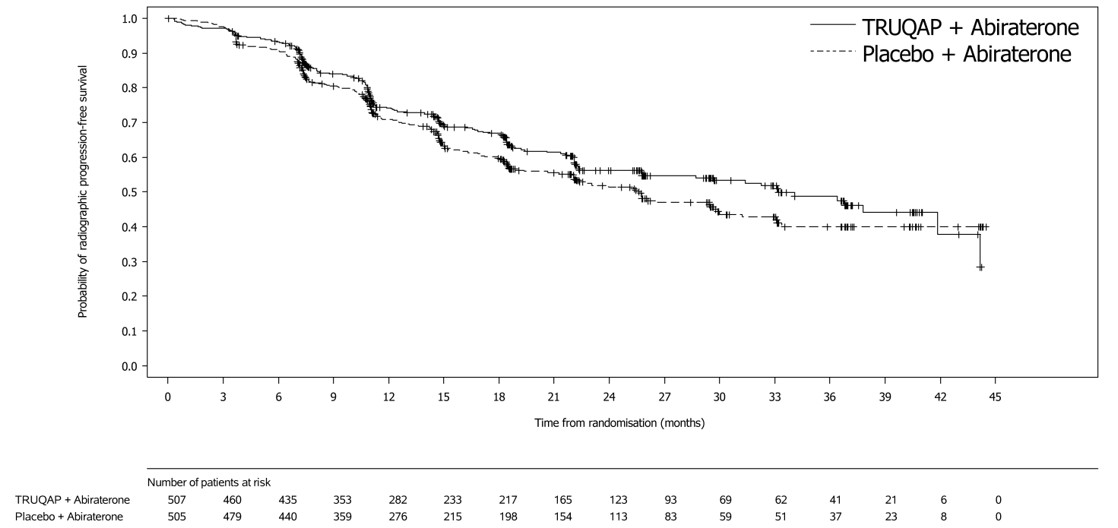
The study demonstrated a statistically significant reduction in the risk of investigator-assessed radiographic progression (rPFS) for patients receiving TRUQAP in combination with abiraterone compared to patients receiving placebo in combination with abiraterone. Results from the blinded independent review committee (BICR) assessment were consistent with the investigator-assessed rPFS results.
Efficacy results are presented in Table 10 and Figure 2. Overall survival results were immature at the time of the rPFS analysis (26% of the patients died).
TRUQAP plus Abiraterone (N=507) | Placebo plus Abiraterone (N=505) | |
Investigator‑Assessed Radiographic Progression-Free Survival (rPFS) | ||
Number of events (%) | 183 (36) | 215 (43) |
Median, months (95% CI) | 33.2 (25.8, 44.2) | 25.7 (22.0, 29.9) |
Hazard ratio (95% CI) Stratified Cox proportional hazards model stratified by volume of disease/visceral metastases, geographic location | 0.81 (0.66, 0.98) | |
p-value Two-sided p-value based on a stratified log-rank test stratified by volume of disease/visceral metastases, geographic location | 0.034 | |
Figure 2: Kaplan-Meier Plot of Radiographic Progression-Free Survival in CAPItello 281
HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
Strength | Description | Package Size and Type | NDC Number |
TRUQAP 160 mg | Beige film-coated, round, biconvex tablets debossed with ‘CAV’ above ‘160’ on one side and plain on the reverse. | HPDE, child resistant, bottle of 64 tablets | 0310-9500-01 |
TRUQAP 200 mg | Beige film-coated, capsule-shaped, biconvex tablets debossed with ‘CAV 200’ on one side and plain on the reverse. | HPDE, child resistant, bottle of 64 tablets | 0310-9501-01 |
TRUQAP 160 mg | Beige film-coated, round, biconvex tablets debossed with ‘CAV’ above ‘160’ on one side and plain on the reverse. | Each carton has 4 blister packs each blister pack containing 16 tablets (total 64 tablets). | 0310-9500-02 |
TRUQAP 200 mg | Beige film-coated, capsule-shaped, biconvex tablets debossed with ‘CAV 200’ on one side and plain on the reverse. | Each carton has 4 blister packs each blister pack containing16 tablets (total 64 tablets). | 0310-9501-02 |
TRUQAP 200 mg | Beige film-coated, capsule-shaped, biconvex tablets debossed with ‘CAV 200’ on one side and plain on the reverse supplied in a blister with a child resistant closure. | Each carton has 4 blister packs with each blister pack containing 8 tablets (total 32 tablets). | 0310-9501-04 |
Storage and Handling
Store TRUQAP at 20°C to 25°C (68°F to 77°F). Excursions permitted to 15°C to 30°C (59°F to 86°F) [see USP Controlled Room Temperature]. Store tablets in the original package. Discard unused tablets after 45 days.
Dispense bottled TRUQAP tablets either in the original bottle or a USP equivalent tight container.
Mechanism of Action
Capivasertib is an inhibitor of all 3 isoforms of serine/threonine kinase AKT (AKT1, AKT2 and AKT3) and inhibits phosphorylation of downstream AKT substrates. AKT activation in tumors is a result of activation of upstream signaling pathways, mutations in AKT1 , loss of phosphatase and tensin homolog (PTEN) function and mutations in the catalytic subunit alpha of phosphatidylinositol 3-kinase ( PIK3CA ).
In vitro , capivasertib reduced growth of breast cancer cell lines including those with relevant PIK3CA or AKT1 mutations or PTEN alteration. In vivo , capivasertib alone and in combination with fulvestrant inhibited tumor growth of mouse xenograft models including estrogen receptor positive breast cancer models with alterations in PIK3CA, AKT1 , and PTEN .
In vitro , capivasertib reduced growth of AR-positive PTEN deficient prostate cancer cell lines. In vivo , capivasertib inhibited tumor growth in mouse xenograft models bearing PTEN-deficient prostate cancer cells and demonstrated increased antitumor activity when used in combination with abiraterone.